Rapid Gene-Drug Interaction Analysis for Personalized NSCLC Therapy Management: A Python-Based Diagnostic Model
Isha Goyal
Grade 10
Presentation
Problem
Lung cancer stands as one of the most pervasive oncological challenges globally, with over 2 million individuals receiving diagnoses in 2020 alone. Among its forms, non-small cell lung cancer (NSCLC) emerges as the predominant subtype, constituting approximately 80-85% of all lung cancer cases in the United States. This year, an estimated 240,000 Americans will be diagnosed with lung cancer, with NSCLC accounting for 188,000 of these cases[1]. Moreover, the prognostics of this disease are poor, with less than 50% of patients surviving beyond the first year, and only 18% surviving beyond the next 5 years, compelling researchers to push the frontiers of current therapeutic models to enhance patient outcomes and longevity.
The majority of NSCLC patients undergo an intensive and invasive treatment regimen, comprising of radiotherapy, chemotherapy, targeted therapy, immunotherapy, or a combination of these. Among these, however, conventional chemotherapies and targeted therapies hold prominence due to their established efficacy, particularly in advanced-stage disease and cases of relapse. Chemotherapy has long been a cornerstone of NSCLC treatment, exerting cytotoxic effects by disrupting DNA replication and cell division. In recent years, targeted therapies have emerged as a significant advancement in NSCLC management, offering more tailored and precise treatment options. Among these, tyrosine kinase inhibitors (TKIs) have garnered significant attention for their ability to target specific molecular alterations; by inhibiting signalling pathways, these agents effectively suppress tumour growth in subsets of NSCLC with certain mutations.
In the ever-evolving landscape of oncology, new advancements in the treatments of NSCLC seem promising in decreasing mortality rates. One leading concern, however, is the emergence of drug-resistant agents, which pose significant obstacles to managing patients with NSCLC. Drug resistance primarily develops during the course of treatment, as cancer cells adapt and evolve in response to drugs. These mutations enable tumours to develop mechanisms that promote resistance, gradually reducing the effectiveness of the therapeutic regimen over time, and necessitating adjustments to treatment strategies to increase overall survival. It is critical to recognize that drug resistance often becomes apparent over a long period, notably shown through diminished response or even tumour progression. However, by the time these symptoms are noticed, valuable treatment opportunities may have been missed, leading to a compromised prognosis and diminished chances of successful intervention. Thus, a prerequisite to tackling drug insensitivity involves the timely detection of specific gene mutations, underscoring the importance of developing rapid and cost-effective diagnostic tools. There exists current research mapping genetic mutations that confer drug resistance, coupled with tools capable of suppressing the expression of these genes. However, bridging the gap between mutation detection and targeted gene suppression necessitates the development of effective methods for scanning patient genomes to identify and flag relevant mutations. As such, this paper aims to address this gap by offering a novel Python-based support system designed to analyze tumour genomes to predict drug resistance in patients with NSCLC.
Method
Research Method:
Inclusion/Exclusion Criteria:
Scanning patients’ genomes is an intense task that entails an abundance of details. When conducting an extensive gene search of this type, the characteristics of variants differ greatly, particularly concerning their roles in cancer progression and treatment resistance. Thus, this project has implemented inclusion and exclusion criteria during the data search phase to ensure the selection of relevant and scientifically validated variants. The selection criteria prioritize variants that have been extensively investigated and well-documented in scientific literature, as well as those cataloged in reputable databases such as ClinVar. Variants must also demonstrate a clear association with resistance to either chemotherapy or targeted therapy (TKIs). Emphasizing the importance of nucleotide-level alterations, the search focused on variants resulting from base changes, including both single nucleotide variants (SNVs) and multiple nucleotide variants (MNVs). Conversely, exclusion criteria served to filter out novel variants lacking sufficient research support and accessibility, as well as variants associated with resistance due to mechanisms such as over/under-expression, RNA transcription (including micro-RNA) and DNA amplification. Furthermore, the scope was restricted to somatic mutations, excluding germline variations associated with inherited resistance.
Data Search & Filtering:
To identify drug-resistant genes, a systematic approach was employed using Pubmed and Google Scholar as primary sources. An initial search was conducted, employing filters and relevant keywords to identify literature reviews exploring potential drug-resistant genes. Subsequently, an initial screening was performed, focusing on abstracts, introductions, and conclusions to identify potential candidates. Genes that were referenced across multiple studies were taken note of as promising contenders for drug resistance. Following that, a full-text screening was conducted, analyzing the mechanisms of drug resistance. Genes employing mechanisms such as over/under expression, amplification, or RNA transcription (including micro-RNAs) to confer drug resistance were excluded. Additionally, those imparting resistance to drugs beyond targeted therapies and chemotherapies were also eliminated from consideration. To determine novelty, the sequences were cross-referenced against Clinvar[2] and Genbank to verify pathogenic response; those absent from either database were classified as novel variants and excluded from the study. Prior to data entry, the final sequences underwent another round of screening, adhering to the aforementioned procedure. Typically, data searches and filtering processes involve multiple assessors to minimize the risk of bias. However, as the screening process was conducted by a single reviewer, multiple screenings were performed to reduce the likelihood of erroneous exclusions or inclusions.
Data Extraction:
Following the filtering process, genomic data was entered into a Google spreadsheet. Extracted information included general details, including the gene name, specific mutation, rs–ID; characteristics, including variant type (SNV, MNV) and drug resistance; genomic coordinates, including chromosomal, base pair, and protein position; as well as mutation details, including original and mutated nucleotides, amino acid change, and molecular consequence. It was also noted if specific drug classes targeted the given gene if applicable.
Computer Model:
The methodology for the identification of potential chemoresistance markers in patient tumour genomes was implemented using the Python programming language, leveraging the BioPython library[3] for genomic processing and analysis. Raw gene data sourced from ClinVar was formatted in FASTA files for computational processing.
Sequence Alignment and Mutation Detection
Sequence alignment and mutation detection are pivotal processes conducted using the Biopython library. Global pairwise alignment is used to align reference and patient sequences. Due to the large size of the sequences, a chunking approach is adopted. Sequences are segmented into manageable chunks for alignment and mutation detection. Through iterative examination of aligned sequences, mutations such as mismatches and gaps are identified. Detected mutations are cataloged and subsequently analyzed to discern their implications in cancer biology.
Sequence Comparison and Nucleotide Detection
To detect mutation, a direct comparison of nucleotide sequences between reference and patient samples is performed. This process enables the detection of mutations by scrutinizing differences in nucleotide composition. By systematically analyzing nucleotide variations, mutations indicative of cancer-associated genomic alterations are identified. The molecular consequences of detected mutations are characterized, as well as potential mechanisms of drug resistance.
Sequence Handling and Input
The methodology entails retrieving reference and patient genomic sequences through FASTA files from Clinvar. Given the large size of the sequences, a chunking strategy is employed to facilitate processing. Sequences are divided into smaller, manageable chunks for alignment and comparison. Standardization of sequence lengths within each chunk is imperative to ensure consistency in alignment and comparison procedures. Additionally, reverse complementation of sequences is applied to capture mutations present on complementary DNA strands (ref. Mechanisms of Drug Resistance)
User Interaction
User interaction is facilitated through a user prompt to specify the gene of interest for mutation testing. Input validation mechanisms are employed to ensure the accuracy of gene selection, thereby optimizing the mutation detection process. The algorithm produced comprehensive reports detailing mutations and predicted drug-resistant markers. Output data included the gene name; mutation name; position; nucleotide base change; variant type; amino acid change; molecular consequence; and drug resistance.
Analysis
Mechanisms of Drug Resistance: [4-18]
From the data search and filtering process (ref. Research Methods), several key genes were identified as strong candidates for drug resistance: EGFR variants T790M, C797S, and D761Y; ALK variants G1202R and L115R; MET variants Y1230C and D1288N; NTRK1 G595R; KRAS G12C, and TP53 R173H. Of these, EGFR, MET and NTRK1 are located on the plus (coding) strand, and TP53, ALK, and KRAS are located on the minus (non-coding). When gene sequencing, the orientation of the strands is crucial; to analyze genes on the minus strand, it is necessary to obtain the reverse complementary sequence before analysis. The majority of these variants are a result of a single-nucleotide polymorphism (SNPs), and confer resistance to targeted therapies, specifically first/second-generation kinase inhibitors; the only exception lies in P53 R173H, which displays chemoresistance.
Table 1. Resistant gene mutations of approved cancer therapies
|
Gene Name |
Target Drugs Clinically Used |
Drug-Resistant Mechanisms |
|
EGFR |
Type I/II EGFR inhibitors: Gefitinib, erlotinib, afatinib, dacomitinib, icotinib, osmertinib |
EGFR T790M |
|
EGFR C797S |
||
|
EGFR D716Y |
||
|
ALK |
Type I/II ALK TKI: crizotinib, ceritinib, brigatinib, alectinib |
ALK G1202R |
|
ALK L1152R |
||
|
MET |
Type I MET TKI: crizotinib, savolitinib, capmatinib |
MET Y1230C |
|
MET D1288N |
||
|
NTRK1 |
Larotrectinib, entrectinib |
NTRK1 G595R |
|
KRAS |
Sotrasib |
KRAS G12C |
|
TP53 |
Doxorubicin, cisplatin, 5-fluorouracil |
TP53 R173H |
EGFR mutations inhibit drug response in NSCLC patients undergoing targeted therapy with TKIs. Notable the T790M mutation renders resistance to first and second-generation EGFR TKIs including gefitinib, erlotinib, and afatinib. The T790M mutation enhances the ATP-binding affinity of EGFR, reducing the efficacy of reversible EGFR TKIs and leading to treatment resistance. Similarly, the C797S mutation confers resistance to both generation TKIs, namely osmertinib, through steric hindrance, blocking the covalent binding of the drug. The D761Y mutation, though less common in clinical settings, is positioned adjacent to residues crucial for forming a salt bridge, facilitating interaction with α- and β-phosphates in ATP binding. This mutation has been linked to acquired resistance to second-generation EGFR inhibitors.
ALK mutations G1202R and L115R emerge as leading resistant-inducing mutations, characterized by their disruption of the ALK kinase domain. The ALK G1202R mutation undermines the efficacy of first and second-generation ALK TKIs, such as crizotinib, ceritinib, alectinib, and brigatinib, by altering the ATP-binding pocket of the ALK domain, impeding optimal drug-receptor interactions. Conversely, the ALK L115R mutation disrupts the structural integrity of the ALK kinase domain, particularly proximal to the ATP-binding site, thereby compromising the binding affinity between ALK TKIs and the ALK protein.
MET mutations Y1230C and D1288N, located within the MET kinase domain, induce constitutive activation of MET signalling by altering the conformation of the kinase domain, promoting continuous downstream signalling pathways crucial for cell proliferation and survival. Moreover, cancer cells bearing Y1230C and D1288N mutations exhibit reduced sensitivity to MET inhibitors, namely Type I TKIs. Furthermore, activation of bypass signalling pathways, including upregulation of alternative receptor tyrosine kinases and downstream effectors, enables cancer cells to sustain growth and survival independent of MET inhibition.
The NTRK1 G595R mutation can confer resistance to targeted therapies through mechanisms rooted in its catalytic domain. This mutation hinders inhibitor binding and potentially augments catalytic function by altering the kinetics of ATP binding. By decreasing the KM for ATP, these mutations intensify the competition between ATP and inhibitor binding, thereby diminishing the effectiveness of targeted therapies designed to disrupt kinase activity. Consequently, cancer cells harbouring the NTRK1 G595R mutation demonstrate a reduced susceptibility to inhibition by NTRK inhibitors, namely larotrectinib and entrectinib.
The KRAS G12C mutation induces a structural modification in the KRAS protein, trapping it in a perpetually active state bound to GTP. As a consequence, downstream signalling pathways crucial for cell proliferation and survival, notably the RAF-MEK-ERK and PI3K-AKT pathways, undergo sustained activation. The RAF-MEK-ERK pathway plays a pivotal role in regulating cell growth, differentiation, and survival by modulating the activity of transcription factors; simultaneously, the PI3K-AKT pathway controls various cellular processes, including metabolism, protein synthesis, and cell survival. The persistent activation of these signalling cascades renders cancer cells housing the KRAS mutation less reliant on external growth signals and renders them resistant to targeted therapies like sotrasib.
TP53, which encodes the p53 transcription factor, is the most frequently altered gene across human tumours. Among its various mutations, TP53 R175H stands out as a significant contributor to chemoresistance in lung carcinoma cells. The mutation promotes the upregulation of miR128-2 through direct binding to the promoter region of its host gene ARPP21. Consequently, miR128-2 suppresses E2F5 expression, thereby reducing the levels of the downstream target gene p21, which exerts an antiapoptotic effect. This miR128-2/E2F5/p21 axis serves as a critical mechanism underlying the resistance of lung carcinoma cells to conventional chemotherapy agents like doxorubicin, cisplatin, and 5-fluorouracil.
Computer Algorithm:
Figure 1. Drug-Resistance Detection Flowchart
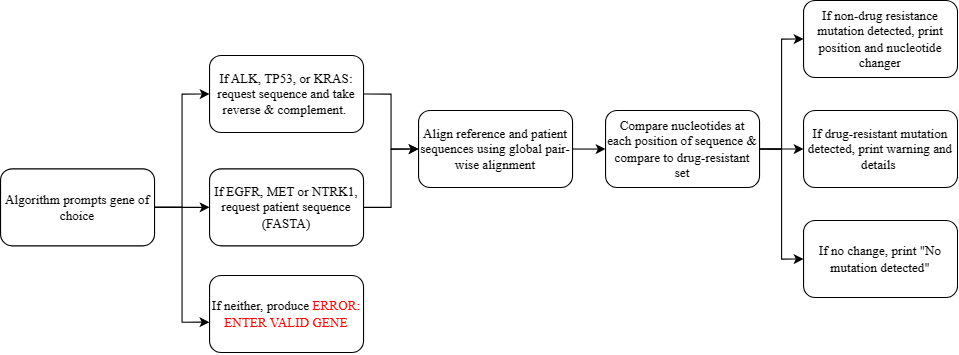
Figure 1 denotes the procedural framework of the algorithm designed for the identification of drug-resistant mutations. Initially, the algorithm prompts the user to select the gene of interest from a predetermined list (EGFR, MET, NTRK1, ALK, KRAS, or TP53). Upon input, if the gene belongs to the former three, the algorithm requests the user to provide the patient sequence in a FASTA[2] format. Conversely, for genes falling within the latter three categories, the algorithm requests the patient sequence and computes the reverse complement of both the patient and reference strands, due to the gene's localization on the minus strand. Failure to provide a valid gene name results in an error message, prompting the user to re-enter the information. After gene selection and sequence acquisition, the algorithm proceeds with a global pair-wise alignment of the patient and reference sequences, identifying gaps indicative of nucleotide mismatches. It then evaluates individual nucleotides at each position, detecting discrepancies between the sequences. Following this comparative analysis, mutations are cross-referenced against a database housing the positional and nucleotide alterations associated with drug-resistant mutations. Upon mutation analysis, the algorithm presents distinct outcomes: if no mutations are detected, the algorithm outputs "no mutation detected"; in case of non-drug resistant mutations, it outputs the position and nucleotide change detected; and upon verification of drug-resistant mutations, the algorithm issues a warning regarding drug resistance, accompanied by detailed information encompassing mutation name, classification, nucleotide variation, amino acid substitution, molecular consequences, and drug resistance implications.
The algorithm was evaluated through manual manipulation of genetic data. While publicly available gene databases exist, they lack the requisite specificity for the targeted genes under investigation. Moreover, these databases lack samples with confirmed chemoresistance, necessitating manual nucleotide changes to verify the functionality and accuracy of the code. Additionally, the testing was constrained to TP53 due to computation limitations; as the other genes are quite large, the algorithm produces a memory error when processing. Three ‘patient sequences’ were tested: the first with no mutation, the second with a non-drug resistant mutation, and the third with a verified drug-resistant modification. The patient sequences were made by copying the reference genome, and making appropriate alterations.
For the first phase of testing, the ‘patient sequence’ received no alterations from the original genome. As such, the following output is given:
Enter the gene you want to test (EGFR, BRAF, NTRK1, MET, ALK, KRAS, or P53): p53
No mutations were detected.
In the second phase of testing, several modifications were made, none of which conferred drug resistance. For instance, one modification changed the final nucleotide from Adenine (A) to Guanine (G); taking the reverse and complement of the sequence, the modification reads from Thymine (T) to Cytosine (C) at position 1. This is not identified as a drug-resistant mutation, and prints as such:
Enter the gene you want to test (EGFR, BRAF, NTRK1, MET, ALK, KRAS, or P53): p53
Modification at position 1: Reference T - Patient C
Multiple mutation detections were also tested, none of which conferred drug resistance. For instance, one modification changed the final nucleotide from Adenine (A) to Guanine (G), and the second-last pair from Cytosine (C) to Adenine (A); taking the reverse and complement, the modification reads from Thymine (T) to Cytosine (C) at position 1, and Guanine (G) to Thymine (T) at position two. It prints as such:
Enter the gene you want to test (EGFR, BRAF, NTRK1, MET, ALK, KRAS, or P53): p53
Modification at position 1: Reference T - Patient C
Modification at position 2: Reference G - Patient T
In the final phase of testing, the algorithm was tested to detect chemoresistant mutations. Due to processing size (ref. Discussion), only TP53 R175H could be tested, which produced the following output:
Chemoresistant mutation detected at index 6, 666.
Variant Name: P53 R173H. Variant Type: SNV.
Mutation: C -> T.
Amino Acid Change: Arginine -> Histamine.
Molecular Consequence: Missense Variant.
Drug Resistance: Doxorubicin, cisplatin, 5-fluorouracil.
Note: Highly pathogenic mutation. Consult doctor.
As such, the algorithm has proven to be successful, with certain limitations to be mentioned in the Discussion section.
Conclusion
Discussion:
To the author’s knowledge, this is the first attempt at a Python-based diagnostic model for drug-resistant diagnosis. The algorithm’s strengths lie in its ability to streamline processes for physicians, offering a more accessible approach to genetic mutation scanning compared to traditional methods involving manual analysis. Moreover, the algorithm's cost-effectiveness is a significant advantage, as it incorporates hard-coded data, thereby reducing the financial burdens associated with targeted therapies and personalized medicine. Additionally, the model's efficiency is commendable, particularly in the context of cancer progression, where timely detection is critical. These developments are particularly pertinent given that less than one-fifth of NSCLC patients survive beyond the 5-year mark, often due to late diagnoses of drug resistance mechanisms. By simplifying the process of inputting genetic information and receiving rapid analysis, the model contributes to improved patient care outcomes.
The project faced several limitations that warrant discussion. Firstly, the diagnostic model's coverage was limited to Single/Multi-Nucleotide Variants (SNV/MNVs), excluding other mutation types such as insertions/deletions, and duplications, as well as mechanisms inducing overexpression and underexpression. Broadening the model's scope to encompass these variants could enhance its clinical relevance and utility. Additionally, due to memory and storage constraints within the operating system, only the TP53 gene could be effectively tested. Future research may benefit from enhanced computing systems to facilitate comprehensive gene testing. Ethical considerations surrounding genetic data handling were also acknowledged, emphasizing the importance of transparent and ethically responsible screening practices. However, since the sequences required for analysis constitute specific segments of tumour DNA, which is already practiced routinely in hospitals, the sequencing methods utilized are familiar and ethically sound. As such, future steps for this project include: (1) refining it to suit different mechanisms conferring drug resistance, (2) upgrading the operating system to work on larger sequences, and, (3) if used in clinical settings, safeguarding patient rights and protecting their genetic data.
Conclusion:
This paper presents a novel Python-based diagnostic model designed for the detection of drug resistance in non-small cell lung cancer (NSCLC) and has successfully developed and tested a prototype. With further refinement and ethical considerations, this innovation has the potential to transform the field of oncology and enhance patient survival rates for the disease
Citations
References:
[1] Cancer.Net. (2023, March). Lung Cancer - Non-Small Cell: Statistics. Cancer.Net. Retrieved February 22, 2024, from https://www.cancer.net/cancer-types/lung-cancer-non-small-cell/statistics
[2]National Center for Biotechnology Information. (2024). ClinVar [Database]. Retrieved from https://www.ncbi.nlm.nih.gov/clinvar/
[3]Peter J. A. Cock, Tiago Antao, Jeffrey T. Chang, Brad A. Chapman, Cymon J. Cox, Andrew Dalke, Iddo Friedberg, Thomas Hamelryck, Frank Kauff, Bartek Wilczynski, Michiel J. L. de Hoon: “Biopython: freely available Python tools for computational molecular biology and bioinformatics”. Bioinformatics 25 (11), 1422–1423 (2009).
[4]Chiang, Y. T., Chien, Y. C., Lin, Y. H., Wu, H. H., Lee, D. F., & Yu, Y. L. (2021). The Function of the Mutant p53-R175H in Cancer. Cancers, 13(16), 4088. https://doi.org/10.3390/cancers13164088
[5]Kryczka, J., Kryczka, J., Czarnecka-Chrebelska, K. H., & Brzeziańska-Lasota, E. (2021). Molecular Mechanisms of Chemoresistance Induced by Cisplatin in NSCLC Cancer Therapy. International journal of molecular sciences, 22(16), 8885. https://doi.org/10.3390/ijms22168885
[6]Lin, J. J., & Shaw, A. T. (2016). Resisting Resistance: Targeted Therapies in Lung Cancer. Trends in cancer, 2(7), 350–364. https://doi.org/10.1016/j.trecan.2016.05.010
[7]Liu, Y. P., Zheng, C. C., Huang, Y. N., He, M. L., Xu, W. W., & Li, B. (2021). Molecular mechanisms of chemo- and radiotherapy resistance and the potential implications for cancer treatment. MedComm, 2(3), 315–340. https://doi.org/10.1002/mco2.55
[8]Lønning, P., Knappskog, S. Mapping genetic alterations causing chemoresistance in cancer: identifying the roads by tracking the drivers. Oncogene 32, 5315–5330 (2013). https://doi.org/10.1038/onc.2013.48
[9]Ramos, A., Sadeghi, S., & Tabatabaeian, H. (2021). Battling Chemoresistance in Cancer: Root Causes and Strategies to Uproot Them. International journal of molecular sciences, 22(17), 9451. https://doi.org/10.3390/ijms22179451
[10]Ranasinghe, R., Mathai, M. L., & Zulli, A. (2022). Cisplatin for cancer therapy and overcoming chemoresistance. Heliyon, 8(9), e10608. https://doi.org/10.1016/j.heliyon.2022.e10608
[11]Souza LCME, Faletti A, Veríssimo CP, Stelling MP, Borges HL. p53 Signaling on Microenvironment and Its Contribution to Tissue Chemoresistance. Membranes (Basel). 2022 Feb 9;12(2):202. doi: 10.3390/membranes12020202. PMID: 35207121; PMCID: PMC8877489.
[12]Sosa Iglesias, V., Giuranno, L., Dubois, L. J., Theys, J., & Vooijs, M. (2018). Drug Resistance in Non-Small Cell Lung Cancer: A Potential for NOTCH Targeting?. Frontiers in oncology, 8, 267. https://doi.org/10.3389/fonc.2018.00267
[13]Wang, X., Zhang, H., & Chen, X. (2019). Drug resistance and combating drug resistance in cancer. Cancer drug resistance (Alhambra, Calif.), 2(2), 141–160. https://doi.org/10.20517/cdr.2019.10
[14]Xiao et al. (2023, February 20). Recent progress in targeted therapy for non-small cell lung cancer. Frontiers. Retrieved February 22, 2024, from https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2023.1125547/full
[15]Yuan, M., Huang, LL., Chen, JH. et al. The emerging treatment landscape of targeted therapy in non-small-cell lung cancer. Sig Transduct Target Ther 4, 61 (2019). https://doi.org/10.1038/s41392-019-0099-9
[16]Young et al. (2023, May 19). Expression of multidrug resistance protein-related genes in lung cancer. Retrieved February 22, 2024, from https://aacrjournals.org/clincancerres/article/5/3/673/199262/Expression-of-Multidrug-Resistance-Protein-related
[17]Zheng H. C. (2017). The molecular mechanisms of chemoresistance in cancers. Oncotarget, 8(35), 59950–59964. https://doi.org/10.18632/oncotarget.19048
[18]Zhou, J., Kang, Y., Chen, L., Wang, H., Liu, J., Zeng, S., & Yu, L. (2020). The Drug-Resistance Mechanisms of Five Platinum-Based Antitumor Agents. Frontiers in pharmacology, 11, 343. https://doi.org/10.3389/fphar.2020.00343
Acknowledgement
This project would not have been possible without the help of my mentors, Dr. Iaci Soares, Mr. Assaf Gordon and Mx. Dallas Mythril, who assisted me in understanding genetic biology and the coding process. I also thank Ms. Manil Bhardwaj at BioAro for providing me with insight into genomic sequencing. Lastly, a deep sense of gratitude goes to the creators of BioPython, a freely available tool for computational molecular biology, as their work was foundational to this project.