Isolates and Characteristics of Genomic Data in Specific Isolates of Pasteurella Bacteria
Shicheng Zhang
Webber Academy
Grade 11
Presentation
No video provided
Problem
With Pasteurella Multocida being a very common AMR and MDR possessing bacteria, measures need to be taken to ascertain the virulence, antibiotic repelling capability, and independent survival mechanisms against foreign intrusion present in the bacteria in general.
With respects to Pasteurella Multocida Isolates PM119, PM125, PM146, PM332, and PM339, using Whole Genome Sequencing (WGS) genomic analysis, can it be determined:
1. What forms of AMR inducing genes are present in each isolate\, what what typical class of antibiotic do the genes in question give the strain of bacteria resistance towards? (e.g: tet _h(3) proteins conducive to Streptomycin resistance)
2. What antiviral defense systems are present in each isolate\, and are there correlations between the number and presence of specific antiviral systems across different isolates? (e.g: Lamassu family antiviral system present in almost all strains\, while defense system PDC-S22 only found in one isolate)
Method
Methodology Section Paper for the Genomic Analysis of Pasteurella Multocida strains PM119, PM125, PM146, PM332, and PM339 in evaluating antiviral resistance, prophage construction, and AMR proteins in the construction of a Phylogenic Tree
Shicheng Zhang1, Sidra Moqaddes2, Dr Dongyan Niu2. 1.Webber Academy, 2.Faculty of Veterinary Medicine, University of Calgary.
Pasteurella is a class of Gram-Negative bacteria belonging to the class of AMR resistant bacterium species, isolated with sensitivity towards penicillin class antibiotics, and subcategorized into serogroups (A, B, D, E, F). Pasteurella results in a large amount of respiratory tract diseases in animals, including cholera in avian mammals and sepsis in cattle. Base species Pasteurella AMR resistance is proven with multiple species. Genomic analysis undertaken focuses on the group of Pasteurella such as the PM119 strain, the PM339 strain, the PM332 strain, the PM125 strain, as well as the PM146 strains, included in the species Pasteurella Multocida.
The mainframe of the analysis of this genomic data comes from the following tools: The FASTP (FASTQ preparation preprocessor), which includes the FASTQC (Quality control processor) to execute quality control on the FASTQ files raw input; The Unicycler program, which disassembles FASTQ raw reads, deletes replicate portions of genetic code, and recombine tangent segments in the genetic code into FASTA files; The Prokka software for the annotation (the identification of certain recognizable segments of genetic code contributing to specific proteins or the creation of systems conducive to AMR and Prophage development, etc.) of completed FASTA files; The quast.py tool (QUAST: Quality Assessment Tool for Genome Assemblies.) for the checking factors such as N50 (continuity of contigs and FASTA files), and similar metrics such as NG50 and NGA50; ABRicate for the screening of contigs (FASTA format) to find specific antibiotics that the bacteria (of which sample was collected and analysed) possesses immunity or resistance towards, as well as specific genomes conducive to such resistance.
Apart from Galaxy, other resources used in this research include: The PADLOC (Prokaryotic Antiviral Defense LOCator) online software developed by the Jackson Lab at the University of Otago, which will be used to specific search for Antiviral Defense systems in the bacteria, including but not limited to CRISPR derivatives.
Stage 1 of the processing for this analysis was done on a local machine. All of the tools installed was installed on Ubuntu 26.04 LTS (resolute raccoon testing developmental branch). Fastp (along with automatic fastqc) and Unicycler were installed manually using the apt package manager. quast.py was installed using pip3 (--break-system-packages option required for PEP 668), and prokka was compiled from source from the official Github repository (https://github.com/tseemann/prokka).
For Fastp processing, the options set was done with options --detect_adapter_for_pe --cut_right, --cut_window_size 4, --cut_mean_quality 20, and --length_required 50. –detect_adapter_for_pe dictating Fastp to overlap the two reads and search them simultaneously, the two FASTQ files being paired reads. –cut_right forces Fastp to search from the 5’ base to 3’ base, or left to right from within the data. –cut_mean_quality 20 to dictate that the Phred running score analysed by Fastqc must not drop below 99% (20 being 99%, 30 being 99.9%, and so forth) to ensure quality control. –length_required 50 sets the finished read to have a length that has to be longer than 50 base-pairs, other wise the fragment will be far too small for any realistic processing. This, and all further processing in Stage 1, was done with a Ryzen 9 9950X CPU with --thread set to 8, using 8 threads out of 32 available threads.
Unicycler was also run locally using --threads set to 8, also using 8 threads. Only required option required was to set --mode normal to make Unicycler form a normal assembly FASTA file. Inside the Unicycler run, spades.py maximum k-mer setting was set to 127. Values tested for adequate k-mer were: 27, 53, 71, 87, 99, 111, 119, 127. Out of 5 isolates runs, 119 was chosen as the most adequate k-mer overlap size for the reassembly of PM119 and PM125, while 111 was chosen as k-mer for PM146, PM332, and PM339. Using k-mer sizes, contigs bridges were formed under repeat resolution, with output shown as (pulled from unicycler.log from PM119 output): Creating SPAdes contig bridges:
Start Path End quality -14 39 → 76 → 44 → 80 → 56 23 26.8 -5 64 24 41.1 23 -56 → 78 → -44 → -75 → -39 -27 26.7
With high quality scores above 20, the quality of the particular reads is shown to be high. Next, bridge application was run by the spades.py program to execute reassembly, with bridging sections in forwards and reverse order:
Applying Bridges:
Bridge type Start → end Path Quality SPAdes -5 → 24 64 41.113 SPAdes -14 → 23 39, 76, 44, 80, 56 26.788 SPAdes 23 → -27 -56, 78, -44, -75, -39 26.748
In particular, path 64 shows iteratives of high overlap with a quality value exceeding 40, indicating that path 64 section of raw contig is a piece which can be plugged in as is to the final contig. The paths of 39 to 56 as well as reverse reads of -56 to -39 show quality values around 26.7, which for a assembly from 5 different contig paths, is more than adequate for assembly.
The final assembled output, for PM119 as a example. had a contig count of 71, with 101 links between each contig in overlap, totaling in the end to 2291677 base pairs, with a N50 quality value of 125371 base pairs, suggesting larger and more complete contigs compared to fragmented and far smaller contigs below the size amounting to 30000 range.
Prokka processing following had options set to –genus Pasteurella and –species multocida, specifying the specific species of bacteria that Prokka needs to identify. Following with the examples centred around PM125, --strain PM125 was also specified for more targeted identification rather than just plain P. multocida in general. --cpus was set to 16 threads during the runtime, utilizing 16 threads for the processing. Output of PM125.txt in the output file settings revealed a combined count of 2114 coding sequences, 48 tRNA sequences, and were found using Prodigal running as a child process within Prokka, with options -m to ignore baseless genome code, -g 11 to specify prokaryote level bacteria targeting, -p single to treat the entire genetic code input as that of one organism rather than differing between “suspected organisms” with genetic code is odd in certain segments, and -f sco to make the output in Simply Coordinate Output (SCO) format to concisely list the start and end of each gene.
The quast.py processing, which is used to further check quality control of all already processed files, was run using options --threads 16, utilizing 16 threads, and --gene-finding, used to allow the marking of contigs with reference data from Genemark.hmm to reference contigs to the formation or proteins. N50 values calculated at 125731 base pairs, while N90 value was 46874. GC base pairs amounted to 40.25%, relatively average for computations with similar bacterial species. With a N value of 0, no gaps were also found in the genome data in general, showing that this particular assembly and corresponding FASTQ input reads were of high quality and possessed little to none intrinsic errors. This output was conducted on that of the FASTA assembly of PM146.
Stage 2 of the processing included the usage of ABRicate, and the PADLOC Defensefinder. These are online tools, although there is a native install available for ABRicate locally on Ubuntu 26.04 (or any stock Linux distribution with good package manager support, and also Windows via download from source or winget if applicable). ABRicate was used as part of the Galaxy Online Bioinformatics Suite (https://usegalaxy.org), while PADLOC was the product of the Jackson Lab at the University of Otago (https://padloc.otago.ac.nz/padloc/).
For ABRicate, the software was run with all default settings within the Galaxy suite, outputting a .TABULAR file output, although it can be opened in regular .txt format. To take the example of species PM339, it was found varying degrees of 99.88% to 100% coverage of the resistant genes of aph class (aph(3’)-la, aph(6)-ld, and aph(3”)-lb), as well as aadA14 traces, which indicate a massive resistance of said species towards Streptomycin class antibiotics.
PADLOC was run without any change in settings or runtime within the Jackson Lab webpage with the tool provided, and returned all traces and identification of antiviral defense systems. Matches against a database and recognising defense systems was done by using HMM references included within the PADLOC web server. Identification of PM332 using PADLOC revealed a large amount of resistant genes belonging to the Lamassu and CAS type resistance families. The PADLOC was run under version 2.0_0, with PADLOCDB version 2.0_0 as well.
Research
Introduction Section Paper on the Genomic Analysis of Different Pasteurella Bacterium Under Defined Sequencing Shicheng Zhang1, Sidra Moqaddes2, Dr Dongyan Niu2. 1.Webber Academy, 2.Faculty of Veterinary Medicine, University of Calgary.
Pasteurella is a class of Gram-Negative bacteria belonging to the class of AMR resistant bacterium species, isolated with sensitivity towards penicillin class antibiotics, and subcategorized into serogroups (A, B, D, E, F). Pasteurella results in a large amount of respiratory tract diseases in animals, including cholera in avian mammals and sepsis in cattle. Base species Pasteurella AMR resistance is proven with multiple species. Genomic analysis we will undertake will focus on the group of Pasteurella such as the PM119 strain, the PM339 strain, the PM332 strain, the PM125 strain, as well as the PM146 strains.
Large-scale comparative genomics utilizing pangenome (genomes found in all strains of a particular species) modeling (typically via tools like Roary or PIRATE) has resolved that P. multocida is divided into two differentiated phylogroups, PmI (Pasteurella Multocida I) and PmII (Pasteurella Multocida II). Carhuaricra et al. (2020) demonstrated that the transition to a pathogenic lifestyle in cattle is driven by metabolic operons, or a cluster of related genes bunched into a mRNA combination, present in a specific niche.¹ Specifically, the trehalose metabolism operon, which regulates the input of the carbohydrate trehalose, and the torCAD system, responsible for trimethylamine N-oxide [TMAO] anaerobic respiration, essential for bacterium to survive under anaerobic conditions, are omnipresent in Phylogroup PmI; such metabolic markers act as "colonization engines" which allow the species to utilize carbon sources straight from the host environment and thrive in the anaerobic, carbohydrate-rich environment of the lower respiratory tract, giving it the ability to far out-reproduce native populations of bacteria during the early stages of Bovine Respiratory Disease (BRD).¹
Standardization of P. multocida genomic data relies on three distinct “levels” of molecular analysis: the cap locus for capsular typing, or to establish the genotype of toolchains known as capsular locuses, essential to the formation of polysaccharides for the cell wall; the waa locus for Lipopolysaccharide (LPS) genotyping, or to determine specific strains of P. multocida directly from the composition of the cell wall and the defense mechanisms of Gram-Negative bacterium such as P. multocida; as well as Multi-Locus Sequence Typing (MLST).² This framework identifies Capsule Type A and LPS Genotype L3 as the definitive "pneumonic profile" for isolates conducive to BRD. By categorizing strains into specific Clonal Complexes (CCs, strains of a bacteria that share a close range common ancestor) using the RIRDC (catalogue of biological reagents to manipulate experiments in determining the genetic composition of bacteria) or multi-host MLST (Multilocus Sequence Typing, isolating bacterial isolates bases off of genetic differentiation) schemes, bioinformaticians can track the global movement of bacterial lineages possessing extreme virulence.² This categorization is essential for establishing a foundation for any de novo (from scratch) assembly, ensuring that newly sequenced isolates are mapped against a known pathome of virulence-associated genes (VAGs, genes directly attributing for reproduction and infection).
In high-density feedlot environments, where cattle is concentrated in small environments and antibiotics are used en masse, genomic surveillance reveals the dominance of specific sequence types of P. multocida, notably ST79 and ST394. A study conducted by researchers Nassar and Alhamami utilized Whole-Genome Sequencing (WGS) to highlight the emergence of ST394 as a major driver of post-mortem lung pathology (presence in lung tissue after cattle death).³ These lineages are characterized by a highly plastic "mobilome," with a high prevalence of Integrative and Conjugative Elements (ICEs, mobile genetic segments found in both Gram-Positive and Gram-Negative bacteria, responsible for constructing transmission mechanisms between cells and bacteria) like ICEPmu1. These elements facilitate the rapid development of multidrug resistance (MDR), transforming historically harmless and ubiquitous strains into highly resistant pathogens that are able to withstand typical antimicrobial interventions used in modern feedlots.³
The structural genomics of ICEPmu-type elements provides the experimental explanation for the propagation of antibiotic resistance. These elements—typically ranging in size from 50 kb to 80 kb (kilobase, 1000 base pairs), consist of conserved modules (modules of genetic data unchanged between prokaryote and eukaryote species) for Integrase (Int, designed to split genetic material to components to the host cell), Excisionase (Xis, regulating when prophages exit the host cell genome and enters the lytic cycle), and Type IV Secretion Systems (T4SS, allowing for transport of proteins and DNA across the cell membrane) for conjugation. A study from Li et al. (2025) emphasized the diversity of “cargo” within these elements, which frequently co-locate AMR genes—such as tet(H), erm(42), and blaROB-1 (Genomes conducive to antibiotic resistance), alongside virulence factors like iron-acquisition operons (operons specific to capturing iron to be used in metabolic processes).⁴ This physical linkage results in co-selection between different strains of P. multocida, where the therapeutic use of one antibiotic (e.g., tetracycline, of which P. multocida is highly resistant towards) aids in maintaining the mobile element (of resistance), including genes for virulence and resistance to unrelated drug classes (eg: penicillin resistance from P. multocida ST394).⁴ Validating bioinformatic predictions against phenotypic data is critical for clinical reliability. A study by Pintér et al. (2025) utilized Broth Microdilution (MIC, used to ascertain the minimal amount of antimicrobial agents required to neutralize microorganisms) to assess the accuracy of genotypic screening via utilizing databases like CARD and ResFinder. While they discovered a close to 1:1 correlation for MIC relating to aminoglycosides and tetracyclines, their study identified a genomic gap in sulfonamide and macrolide (both classes of specific antibiotics) resistance.⁵ Several isolates exhibited phenotypic resistance despite the absence of known resistance triggers like sul2 or erm(42). This suggests that P. multocida possesses uncharacterized resistance mechanisms, such as previously undiscovered efflux pumps (protein pump that excretes unwanted substances from cells) or point mutations in ribosomal proteins (the altering of a nucleotide base to change the properties of the protein produced by the ribosome), that existing research methods are not prone to recognising as of yet.⁵
Regarding the genetic composition of P. multocida itself, complete genome assembly of isolates possessing enhanced virulence capacity (e.g., Strain P6) reveals a composition of approximately 2.29 Mb (mega base pairs) with a 40.2 GC (Guanine-Cytosine Pairs) content. Zhao et al. (2024) identified multiple prophages and Genomic Islands (GIs, genomes and segments of genes acquired through transferring between different bacteria and/or viruses) that serve as reservoirs for potentially toxic cargo genes.⁶ Moreover, the presence of a Type II-C CRISPR-Cas ((A class of CRISPR system that utilises only the Cas9 Cas effector), (CRISPR: Immune system like response and intruder removal mechanisms for prokaryotic bacteria, heavily contributes to AMR)) system provides these pathogens with a selective immune system; it prevents the "toxic" integration of bacteriophages while allowing the preservation of "useful" prophages that enhance the ability for host colonization. This balance of genomic stability and horizontal acquisition is a cardinal advantage of successful P. multocida pathogens.⁶
The genetic potential of certain infectious agents is ultimately executed through gene expression (Producing a protein or similar functional compound from information encoded by DNA/RNA), and RNA-Seq analysis has revealed a significant "fitness cost" associated with enhanced levels of antimicrobial resistance. Zhan et al. (2021) demonstrated that the acquisition of fluoroquinolone (broad spectrum antibiotic) resistance leads to the downregulation (decreased production) of over 1,100 genes, including the Capsular Polysaccharide (CPS, crucial for external interactions of the bacteria and virulence) synthesis cluster and iron-uptake systems.⁷ This rewiring results in attenuation, where the bacteria survive drug exposure but likewise their ability to evade host phagocytes or intake iron is vastly reduced. This phenomenon is critical for why genomic surveys sometimes identify resistant strains that fail to cause acute disease in experimental situations.⁷
Case-control pangenome (cross-species related genomes) analysis of healthy versus BRD-affected cattle indicates that pathogenicity is not strictly related to the presence of certain Virulence-Associated Genes (VAGs). Research by Garzon et al. (2025) found that the distribution of "core" and "accessory" genes, modeled by Heaps' Law (a relation between distinct elements in a array to the total length of the array) as an "open" pangenome, did not significantly differ between commensal (harmless to humans) and pathogenic isolates.⁸ This suggests that BRD is an mostly opportunistic pathology where the host's physiological state and simultaneous infections from different intruders act as primary triggers for the "activation" of dormant genomic potential, rather than the introduction of a genetically distinct "pathotype" of bacteria.⁸
The zoonotic potential of P. multocida is often belittled by the high genomic similarity between livestock strains and those causing human infections. Smallman et al. (2024) highlighted that subspecies septica (P. septica) isolates recovered from human wounds often harbor the same L3 LPS genotype found in bovine BRD.⁹ This source introduced PastyVRDB, a manually isolated database that captures species-specific variants of adhesins (attachment proteins for latching to surfaces) and toxins. Utilizing this tool allows for the identification of markers showing particular host-adaptation, such as specific L-fucose (sugar which aids in transmission of signals within cells) uptake pathways, which facilitate the transition of the pathogen between mammalian species.⁹
With pertinence to prevention and potentially drugs to serve as potential vaccines against infection from P. multocida, the failure of traditional bacterin vaccines to provide cross-protection has led to the emergence of Reverse Vaccinology (RV). Tesfaye et al. (2025) reviewed the use of bioinformatic pipelines (e.g., Vaxign) to screen the core genome for Outer Membrane Proteins (OMPs) that exhibit high antigenicity (to bind to immune system components) and low variability.¹⁰ Candidates such as OmpH, PlpE, and VacJ are prioritized based on predicted MHC-I/II (Major Histocompatibility Complex, a group of genes which serves to encode the proteins on the cellular surface) binding affinity and subcellular localization. This "genome-to-vaccine" approach aims to develop platforms that target conserved epitopes (antigens easily recognizable by the immune system), providing a broad-spectrum solution that remains effective regardless of the shifting evolution or the high degree of LPS diversity found in global P. multocida populations.¹⁰
All pertaining information on the Pasteurella multocida pangenome provides a critical bioinformatic framework for interpreting the genomic architecture of isolates 119, 125, 146, 332, 339, and PM. By synthesizing and recognising the global phylogenetic markers defined by the study Carhuaricra et al. (2020)¹ with the feedlot-specific virulence profiles identified by Alhamami et al. (2023)³, the analysis of these specific isolates will move to a granular assessment of their pathogenic potential. Specifically, this study will systematically interrogate these six genomes for high-risk Sequence Type backgrounds and the Capsule signature (protective external layer), while simultaneously mapping their evolution for ICEPmu-type integrative elements that drive MDR. Furthermore, by utilizing tools similar to PastyVRDB⁹ to screen for metabolic adaptations and conserved outer membrane proteins, this research aims to determine whether isolates 119, 125, 146, 332, and 339 represent commensal residents or hyper-virulent, vaccine-resistant lineages capable of driving acute bovine respiratory disease in cattle and/or humans.
References
- Carhuaricra D, Espinoza-Culupú A, Ramos-García O, et al. Global phylogeny and metabolic adaptation of Pasteurella multocida through pangenome analysis. Sci Rep. 2020;10:13673.
- Wilson BA, Ho M. Pasteurella multocida: Genotypes and Genomics. In: Pasteurella multocida. Current Topics in Microbiology and Immunology. Springer; 2013/2019.
- Alhamami T, Nassar M, et al. Genomic profiling of Pasteurella multocida isolated from feedlot cases of bovine respiratory disease. Vet Microbiol. 2023;284:109825.
- Li Y, et al. Antimicrobial Resistance in Pasteurella multocida Isolates from Bovine Mastitis Can Be Associated with Multidrug-Resistance-Mediating Integrative and Conjugative Elements (ICEs). J Anim Sci. 2025.
- Pintér N, et al. Comparative Analysis of Phenotypic and Genotypic Antibiotic Susceptibility of Pasteurella multocida Isolated from Various Host Species in France and Hungary. Antibiotics. 2025;14(1):68.
- Zhao L, et al. Whole-Genome Sequencing and Pathogenic Characterization of a Pasteurella multocida Serotype A Isolate from a Case of Respiratory Disease. Pathogens. 2024;13(4):312.
- Zhan Y, et al. Genomic and Transcriptomic Analysis of Bovine Pasteurella multocida Serogroup A Strain Reveals Insights Into Virulence Attenuation. Front Vet Sci. 2021;8:707254.
- Garzon A, et al. Comparison of virulence and resistance genes in Mannheimia haemolytica and Pasteurella multocida from dairy cattle with and without bovine respiratory disease. Vet Microbiol. 2025;301:109968.
- Smallman MB, et al. Pathogenomic analysis and characterization of Pasteurella multocida strains recovered from human infections. Emerg Microbes Infect. 2024;13(1):234567.
- Tesfaye T, et al. Advances in Pasteurella multocida Vaccine Development: From Conventional to Next-Generation Strategies. Vaccines. 2025;13(2):142.
Data
A analysis of processed data resulting the AMR resistance and presence of antiviral defense systems concurred the following:
PM119: AMR Genes: tet_h (3), aadA14_1 Antibiotics this strain is resistance towards: Streptomycin, Tetracycline, Doxycycline
Antiviral Defense Systems: Lamassu, GAO-19, PDC-S07, PDC-S23, CRISPR-ARRAY, PDC-M11, AbiD, cas_type_I-F1, dXTPase Targets: Bacteriophages, Plasmids DNA segments, Conjugative Elements (ICEs) System Types: Adaptive Immunity, Innate PDC, Phage Replication Cycle, Cell Suicide to prevent spread
PM125: AMR Genes: None Antibiotics resistant towards: None shown
Antiviral Defense Systems: PDC-S07, PDC-S23, CRISPR-ARRAY, cas_type_I-F1, dXTPase, AVAST_type_II, PDC-S22, RM_type_I, PDC-S15, PDC-M66, ietAS, PDC-S05 Targets: Bacteriophages, Plasmid DNA segments, ICEs System Types: Adaptive Immunity, Innate PDC, Phage Replication Cycle, Foreign DNA deletion Note: Lacks Systems present in other strains, Many systems suggest different environment or high external infection pressure.
PM146: AMR Genes: aad_A14_1, aph (3")-lb_2, aph (3')-la_3, aph (6)-ld_1 Antibiotics resistanct towards: Streptomycin
Antiviral Defense Systems: Lamassu, GAO-19, PDC-S07, PDC-S23, CRISPR-ARRAY, PDC-M11, AbiD, cas_type_I-F1, dXTPase, cas_type_other Targets: Bacteriophages, Plasmid DNA segments, ICEs System Types: Adaptive Immunity, Innate PDC, Phage Replication Cycle, Cell Suicide to prevent spread
PM332: AMR Genes: None Antibiotics resistant to: None shown
Antiviral Defense Systems: Lamassu, GAO-19, PDC-S07, PDC-S23, CRISPR-ARRAY, PDC-M11, AbiD, cas_type_I-F1, dXTPase Targets: Bacteriophages, Plasmids DNA segments, Conjugative Elements (ICEs) System Types: Adaptive Immunity, Innate PDC, Phage Replication Cycle, Cell Suicide to prevent spread
PM339: AMR Genes: aad_A14_1, aph (3")-lb_2, aph (3')-la_3, aph (6)-ld_1 Antibiotics resistanct towards: Streptomycin
Antiviral Defense Systems: Lamassu, GAO-19, PDC-S07, PDC-S23, CRISPR-ARRAY, PDC-M11, AbiD, cas_type_I-F1, dXTPase Targets: Bacteriophages, Plasmids DNA segments, Conjugative Elements (ICEs) System Types: Adaptive Immunity, Innate PDC, Phage Replication Cycle, Cell Suicide to prevent spread
Conclusion
A analysis of processed data resulting the AMR resistance and presence of antiviral defense systems concurred the following:
PM119: AMR Genes: tet_h (3), aadA14_1 Antibiotics this strain is resistance towards: Streptomycin, Tetracycline, Doxycycline
Antiviral Defense Systems: Lamassu, GAO-19, PDC-S07, PDC-S23, CRISPR-ARRAY, PDC-M11, AbiD, cas_type_I-F1, dXTPase Targets: Bacteriophages, Plasmids DNA segments, Conjugative Elements (ICEs) System Types: Adaptive Immunity, Innate PDC, Phage Replication Cycle, Cell Suicide to prevent spread
PM125: AMR Genes: None Antibiotics resistant towards: None shown
Antiviral Defense Systems: PDC-S07, PDC-S23, CRISPR-ARRAY, cas_type_I-F1, dXTPase, AVAST_type_II, PDC-S22, RM_type_I, PDC-S15, PDC-M66, ietAS, PDC-S05 Targets: Bacteriophages, Plasmid DNA segments, ICEs System Types: Adaptive Immunity, Innate PDC, Phage Replication Cycle, Foreign DNA deletion Note: Lacks Systems present in other strains, Many systems suggest different environment or high external infection pressure.
PM146: AMR Genes: aad_A14_1, aph (3")-lb_2, aph (3')-la_3, aph (6)-ld_1 Antibiotics resistanct towards: Streptomycin
Antiviral Defense Systems: Lamassu, GAO-19, PDC-S07, PDC-S23, CRISPR-ARRAY, PDC-M11, AbiD, cas_type_I-F1, dXTPase, cas_type_other Targets: Bacteriophages, Plasmid DNA segments, ICEs System Types: Adaptive Immunity, Innate PDC, Phage Replication Cycle, Cell Suicide to prevent spread
PM332: AMR Genes: None Antibiotics resistant to: None shown
Antiviral Defense Systems: Lamassu, GAO-19, PDC-S07, PDC-S23, CRISPR-ARRAY, PDC-M11, AbiD, cas_type_I-F1, dXTPase Targets: Bacteriophages, Plasmids DNA segments, Conjugative Elements (ICEs) System Types: Adaptive Immunity, Innate PDC, Phage Replication Cycle, Cell Suicide to prevent spread
PM339: AMR Genes: aad_A14_1, aph (3")-lb_2, aph (3')-la_3, aph (6)-ld_1 Antibiotics resistanct towards: Streptomycin
Antiviral Defense Systems: Lamassu, GAO-19, PDC-S07, PDC-S23, CRISPR-ARRAY, PDC-M11, AbiD, cas_type_I-F1, dXTPase Targets: Bacteriophages, Plasmids DNA segments, Conjugative Elements (ICEs) System Types: Adaptive Immunity, Innate PDC, Phage Replication Cycle, Cell Suicide to prevent spread
With respects to AMR and MDR, the following can be ascertained.
The total antibiotics that the sampled isolates bore resistance towards are Streptomycin, Tetracycline, and Doxycycline. Tetracycline is commonly used in cattle feedlots, prime breeding ground for Pasteurella Multocida in general, Streptomycin is a commonly serious case infection antibiotic for Mycobacterium classes, including tuberculosis inducing bacterial types. Doxycycline is a specific branch of Tetracycline designed to treat a wide range of small ailments such as persistent acne. In particular, the presence of resistance to Streptomycin in 3 isolates indicates growing resistance from Pasteurella Multocida isolates towards last resort antibiotics, highlighting the need to diversify the categories of antibiotic classes used and the need to less antibiotic reliant bacterial treatment methods such as the progression of phage treatment.
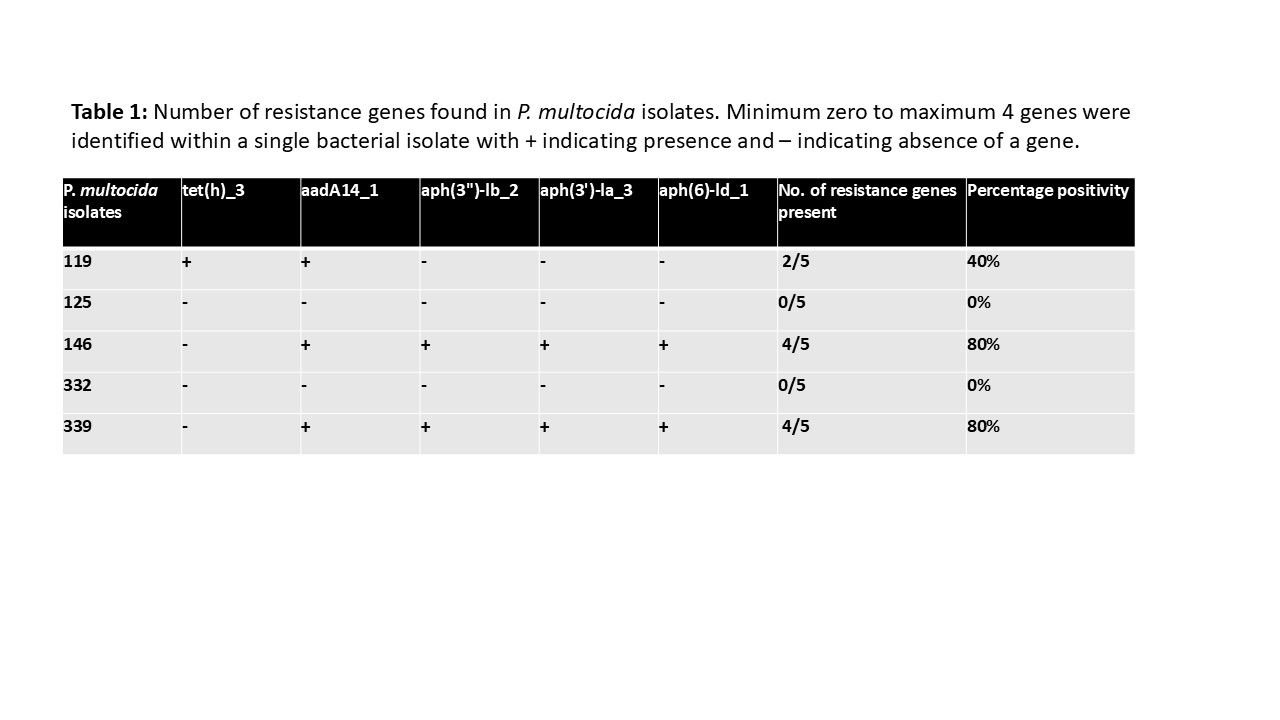
Genes conducive to resistance detected are: - tet(h)_3 - aadA14_1 - aph (3")-lb_2 - aph (3')-la_3 - aph (6)-ld_1
Out of these genes, the tet(h)_3 genome encodes a tetracycline pump to actively remove tetracycline antibiotics from the bacteria. aadA14_1 is a aminoglycoside modification genome which modifies aminoglycoside antibiotics, which includes streptomycin. aph (3")-lb_2, aph (3')-la_3, and aph (6)-ld_1 are all phosphorylate proteins to aminoglycoside antibiotics, preventing their ability to bind to ribosomes and use ribosomes inside infected bacteria to reproduce more of the antibiotic. All of the aad and aph antibiotics explicitly results in streptomycin support, since streptomycin is a aminoglycoside antibiotic.
PM119 possesses genes tet (h)_3, and aadA14_1. This gives is resistance to all 3 antibiotics registered in the resistance measuring: Streptomycin, Tetracycline, and Doxycycline. tet (h)_3, while mainly against tetracycline, offers partial resistance towards Doxycycline as well based on it's composition as a second generation tetracycline class antibiotic. aadA14_1 offers up combined Streptomycin resistance.
PM125 and PM332 registered no antibiotic resistance genomes, and thus no antibiotics resistant to was registered. However, it is crucial to note that the compendium of AMR genomes is not yet fully catalogued, and there may be resistance to certain types of chemicals within both isolates that have yet to be discovered or identified as a particular mechanism.
PM146 and PM339 shared presence of aadA14_1, aph (3")-lb_2, aph (3')-la_3, and aph (6)-ld_1. This combination of aad and aph genomes gives PM146 and PM339 unparalleled aminoglycoside and conversely streptomycin resistance.
With respects to antiviral systems, the following can be ascertained.
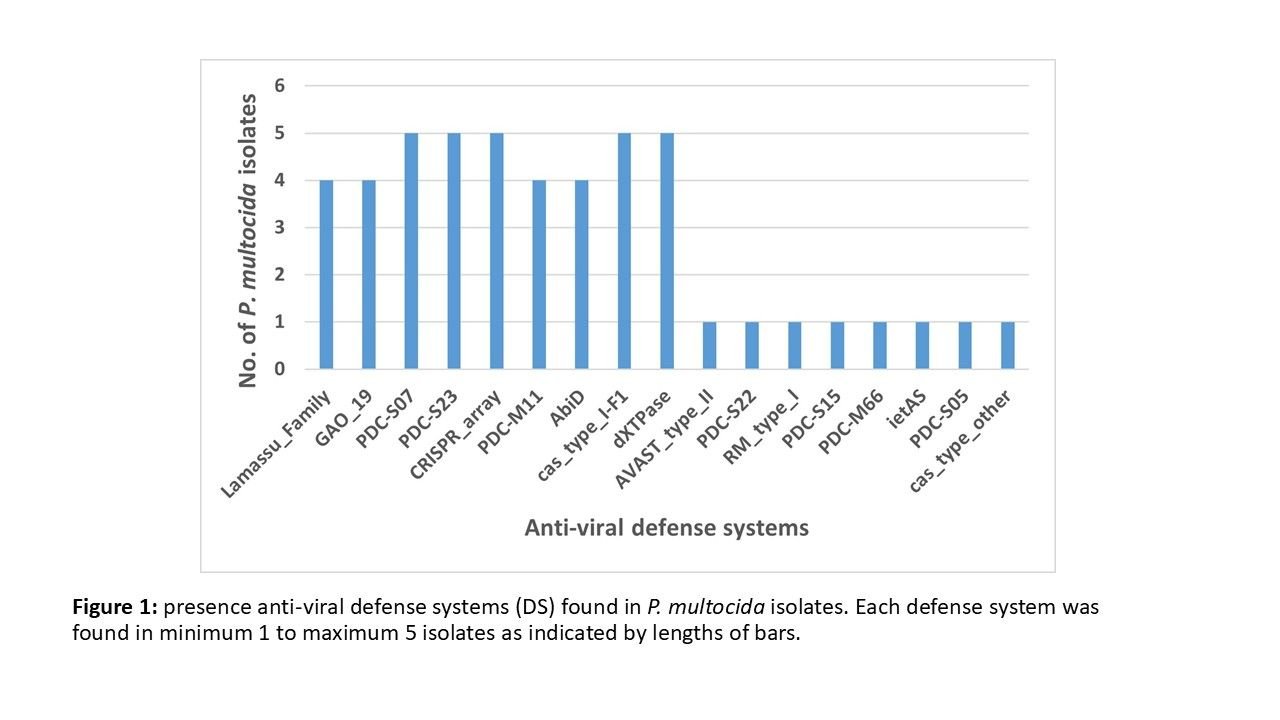
The total of antiviral systems ascertained are: Lamassu Family System. Gao Type 19. PDC Variants S05, S07, S15, S22, S23, M11, M66. CRISPR Array. AbiD. Cas systems types I-F1 and a other unidentified type. dXTPase. AVAST Type 2, RM Type 1 System, and ietAS. - Lamassu Family Systems: Defends against phages by employing a abortive infection suicide method to result in cellular death upon prophage injection. - Gao Type 19 (GAO_19): Poorly categorised system that is recently discovered\, but is proven to target phages. - PDC S-Series: Short for Prokaryotic Defense Cluster\, PDC S-Series systems detect the replication of prophage genetic material\, and interfere with the transfer of replicated material as well as the reproduction sequence. PDC S-Series also contains a abortive infection method. - PDC M-Series: M-Series PDC systems scramble the metabolic processes of the bacterium being infected\, eliminating resources for phage replication. PDC M-Series is linked commonly with abortive infection mechanisms like Lamassu. - CRISPR Array: CRISPR is a adaptive immunity system\, similar to the actual immune system in multicellular organisms. CRISPR preserve "spacers" of DNA from previous invading agents\, inetgrate them with guiding RNA strands\, and use these RNA strands as markers for Cas proteins to cut and neutralise upon infection. This is similar to the antibody system in human immune systems. - Cas system type I-F1 (cas_type_I-F1): This particular Cas type system is a guidance complex which guides Cas3 type proteins to phage tissue and prophages\, facilitating neutralisation. - Cas system type other: A unknown Cas variant. - AbiD: Triggers host cell death when phage gene expression is detected. - RM Type 1 System: Short for Restrictive-Modification systems type 1\, Host DNA with this system is "methylated" and identified\, whilst extraneous and external DNA injected\, without the methylate mark\, is cut and neutralised. - dXTPase: Targeting Phages\, dXTPase is a system designed for the degradation of existing dNTPS in the cellular tissue\, making it impossible for the phages to recombine injected prophage DNA. - AVAST Type 2: Abortive infection mechanism by activating a toxic internal chemical release when phage protein activity is registered (similar to cytokine storm in humans) - ietAS: Likely targeted towards phages\, this is a defense system aimed at preventing gene duplication (transcription inference) by disrupting the RNA polymerase (enzyme responsible for transcription)\, therefore making the phage prophage impossible to replicate.
PM119, PM146, PM332, and PM339 all possess the systems Lamassu, Gao Type 19, PDC S07, S23, CRISPR Array, PDC M11, AbiD, cas_type_I-F1, and dXTPase. PM146 does have an extra cas_type_other system, but relatively, the antiviral defense composition of these 4 isolates remain relatively equal. Looking at this particular pool, it can be ascertained a number of abortive infection systems (e.g: Lamassu in all), replication inference system (e.g: PDC S-Series systems), and active infection resistance (Cas types). This suggests that these isolates possess a well balanced array of antiviral defense systems for use against phages and other external antigens such as heavy metals (particularly with active defense systems such as Cas which can bind to chemicals and materials as well as specific genes).
PM125, however, possess a far different and far more diversified series of antiviral defense systems. Immediately, Lamassu, Gao Type 19, PDC M11, and AbiD, systems present in all other systems, are not present within PM125. Upon closer inspection, it can be further noted that 3 out of the 4 missing systems share common targeting towards phages. Upon first glance, it can be inferred from this change that PM125 is not specified for phage defense. However, this is not the case, since a multitude more antiviral defense systems are present, such as AVAST, ietAS, and more PDC S-Series systems. The presence of so many PDC S-Series systems signals that PM125 is a isolate that either possessed former evolutionary vulnerability towards phages or thrives in a environment replete with natural phage content, therefore needing to develop and maintain such immunity. Furthermore, the lack of some newer systems such as Gao Type 19 but the presence of other new defense systems such as AVAST signals that PM125 is developing on a different evolutionary path than that of the other 4 isolates sampled and analysed. The lack of the four previously isolated defense systems can also be explained individually: PDC-M11 may have been replaced by PDC-M66, AbiD may have been proven genetically obsolete by the development of AVAST, the same being potentially said for Lamassu. Gao Type 19 is also a relatively newly identified system, which potentially signifies that PM125 evolving with AVAST is moving on a different evolutionary trend than that of the 4 other isolates.
In combination, analysis for the 5 isolates is as follows.
PM119: Shares the common defense system isolates with 146, 332, and 339. Possesses greater antibiotic resistance with resistance to Streptomycin, Tetracycline, and Doxycycline. This suggests that in terms of phages and antiviral, 119 possesses a typical, balanced antiviral system congregation, but it possessed the largest of all antibiotic resistance out of the 5 isolates suggesting prolonged exposure to aminoglycoside antibiotics employed in feedlots, as well as potentially large amounts of exposure to tetracycline to also develop associated second generation doxycycline resistance.
PM125: Possesses no antibiotic resistance but harbours the most unique set of antiviral defense systems seen in all 5 isolates. This suggests prolonged exposure to different types of phage or other virus types externally, while quite limited in exposure to antibiotics.
PM146: Possesses antibiotic resistance towards streptomycin, suggesting nominal aminoglycoside resistance, as well as balanced amount of antiviral defenses, like all 4 isolates apart from PM125. However, it does have the cas_type_other evolution, suggesting a potential beginning of further evolution in terms of antiviral defense systems in comparison to the other isolates.
PM332: Possesses no antibiotic resistance, as well as equal antiviral defense systems as PM119 and PM146 without the cas_type_other system in PM146, suggesting nominal defenses and adaption to viruses and phages.
PM339: Possesses antibiotic resistance towards streptomycin, suggesting nominal aminoglycoside resistance. Antiviral systems contain the same pertinent amount of systems as 119, 146 (without cas_type_other), 332, and 339.
In summation with combined research from my mentor into MIC testing, the following can be ascertained:
The analysis of antiviral defense systems and antimicrobial resistance in the 5 isolates concluded that:
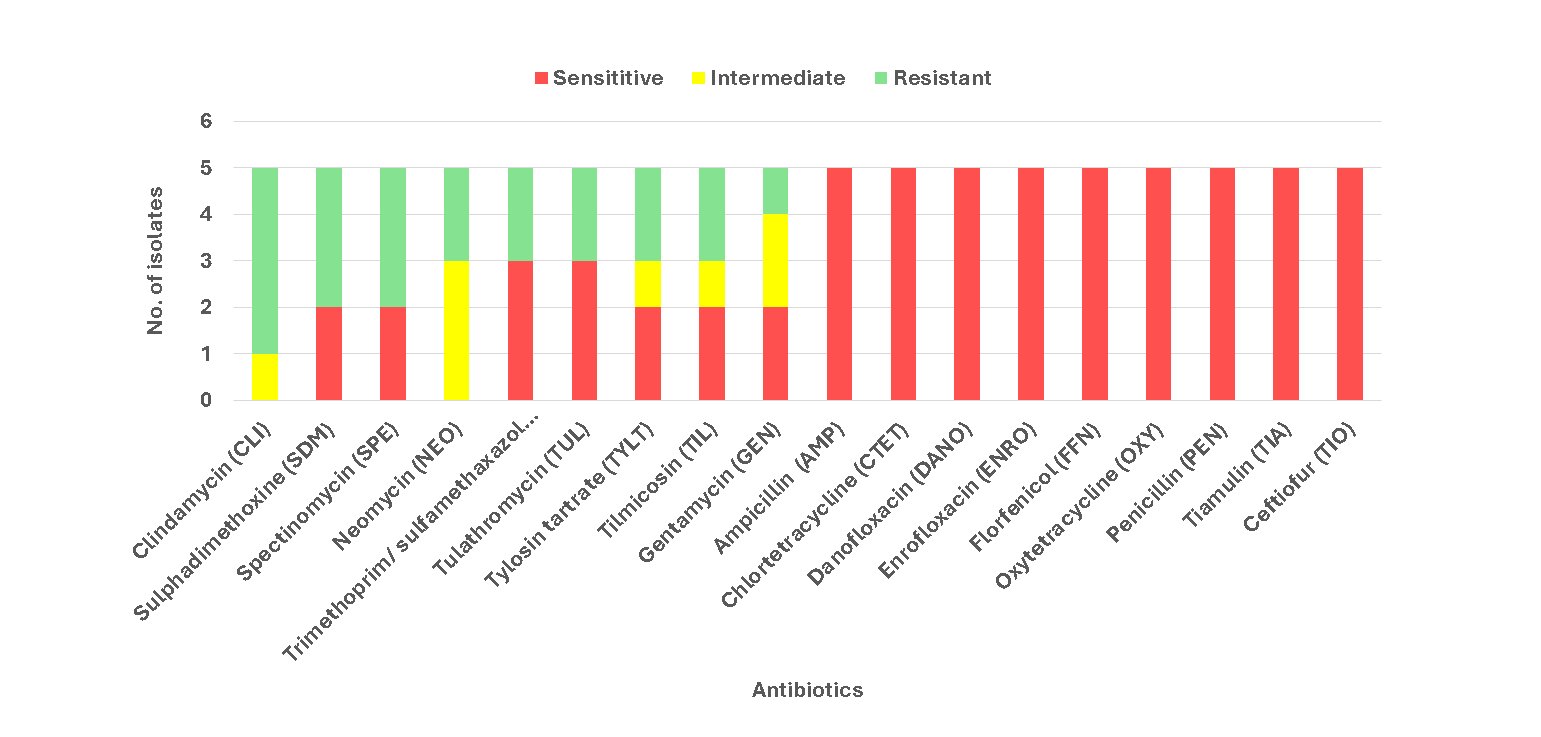
1. AMR genes aad and aph class were detected, giving resistance towards aminoglycoside1 class antibiotics, such as Gentamicin, Neomycin, and Streptomycin, found in three isolates (PM119, PM146, and PM339).
2. AMR gene tet (h)_3 was also detected in PM119. However, given sensitivity was noted from all 5 isolates towards tetracyclines, this at most indicates developing resistance towards tetracyclines.
3. Isolates on average possessed resistance towards Aminoglycosides, Macrolides, and Lincosamide antibiotics.
4. Four isolates possessed Lamassu family2, PDC S-Series3 system, and CRISPR/Cas type antiviral defense systems. The makeup of individual antiviral defense systems was near identical across four strains (PM119, PM146, PM332, and PM339).
5. PM125 contained seven other distinct antiviral defense systems in comparison to the other four strains sampled. It also lacks four abortive infection and metabolic influence antiviral defense systems found in the 4 other strains.
Citations
- GALAXY
Galaxy. Galaxy. Usegalaxy.org. Published 2019. https://usegalaxy.org/ 2. PADLOC Jackson Lab Bioinformatics Server. Otago.ac.nz. Published 2022. https://padloc.otago.ac.nz/padloc/ 3. PHASTEST David S. Wishart, Scott Han, Sukanta Saha, Eponine Oler, Harrison Peters Jason Grant, Paul Stothard, Vasuk Gautam (2023), PHASTEST: Faster than PHASTER, Better than PHAST, Nucleic Acids Research (Web Server Issue), https://doi.org/10.1093/nar/gkad382
6. Prokka Seemann T. Prokka: rapid prokaryotic genome annotation Bioinformatics 2014 Jul 15;30(14):2068-9. PMID:24642063 DOI:10.1093/bioinformatics/btu153 CFF
7: FastQC Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data [Online]. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
8: Unicycler Wick RR, Judd LM, Gorrie CL, Holt KE. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol. 2017;13(6):e1005595. doi:10.1371/journal.pcbi.1005595
9. QUAST Mikheenko, A., Berard, A., Gurevich, A., et al. (2023). QUAST: quality assessment tool for genome assemblies. Nucleic Acids Research, 51(W1), W601–W606. https://doi.org/10.1093/nar/gkad406
10. FASTP Chen, S., Zhou, Y., Chen, Y., & Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics, 34(17), i884-i890. doi: 10.1093/bioinformatics/bty560.
- Carhuaricra D, Espinoza-Culupú A, Ramos-García O, et al. Global phylogeny and metabolic adaptation of Pasteurella multocida through pangenome analysis. Sci Rep. 2020;10:13673.
- Wilson BA, Ho M. Pasteurella multocida: Genotypes and Genomics. In: Pasteurella multocida. Current Topics in Microbiology and Immunology. Springer; 2013/2019.
- Alhamami T, Nassar M, et al. Genomic profiling of Pasteurella multocida isolated from feedlot cases of bovine respiratory disease. Vet Microbiol. 2023;284:109825.
- Li Y, et al. Antimicrobial Resistance in Pasteurella multocida Isolates from Bovine Mastitis Can Be Associated with Multidrug-Resistance-Mediating Integrative and Conjugative Elements (ICEs). J Anim Sci. 2025.
- Pintér N, et al. Comparative Analysis of Phenotypic and Genotypic Antibiotic Susceptibility of Pasteurella multocida Isolated from Various Host Species in France and Hungary. Antibiotics. 2025;14(1):68.
- Zhao L, et al. Whole-Genome Sequencing and Pathogenic Characterization of a Pasteurella multocida Serotype A Isolate from a Case of Respiratory Disease. Pathogens. 2024;13(4):312.
- Zhan Y, et al. Genomic and Transcriptomic Analysis of Bovine Pasteurella multocida Serogroup A Strain Reveals Insights Into Virulence Attenuation. Front Vet Sci. 2021;8:707254.
- Garzon A, et al. Comparison of virulence and resistance genes in Mannheimia haemolytica and Pasteurella multocida from dairy cattle with and without bovine respiratory disease. Vet Microbiol. 2025;301:109968.
- Smallman MB, et al. Pathogenomic analysis and characterization of Pasteurella multocida strains recovered from human infections. Emerg Microbes Infect. 2024;13(1):234567.
- Tesfaye T, et al. Advances in Pasteurella multocida Vaccine Development: From Conventional to Next-Generation Strategies. Vaccines. 2025;13(2):142.
Acknowledgement
All of the research that I conducted was conducted under the guidance and teaching of:
Ms. Sidra Moqaddes, Faculty of Veterinary Medicine, University of Calgary
Ms. Mawra Gohar, Faculty of Veterinary Medicine, University of Calgary
Dr. Dongyan Niu, Faculty of Veterinary Medicine, University of Calgary
The Faculty of Veterinary Medicine, University of Calgary
and Dr. Beatriz Garcia-Diaz, Webber Academy.
Acknowledgement of software and sources that I utilised can be found in the Citations Segment of the Portal