
Investigating lysosomal storage disease mucopolysaccharidosis(III) and viral vector gene therapy treatments
Mia Davison
STEM Innovation Academy High School
Grade 11
Presentation
No video provided
Problem
Brief Overview: Mucopolysaccharidosis is a lysosomal storage disease where genetic mutations cause for a lack of a glycosaminoglycan enzymes to be produced. This leads to inflammation from over-signaling cytokines resulting in neurodegeneration and somatic function complications. MPS consists of seven types, each varying in severity.
Epidemiology & Statistics: Studies in the US were able to pull data from 789 patients throughout 1995-2015 (Puckett Y, et al, 2021). Prevalence throughout all types was averaged 2.67 per million, classifying it as a rare disease. However take into account US population estimated over 300 million (varying throughout years of data collection). Incidence of MPS was more common in type I, II, & 3, which is also reflected globally (Celik B, et al, 2021; Puckett Y, et al, 2021). According the the Canadian MPS society, about 1 in 25000 children suffer from this disease within Canada. Another study was done across 33 countries and it was found Saudi Arabia had one of the highest prevalence rates estimated to be due to low genetic variation within the population (or the "founder effect") (Celik B, et al, 2021). MPSIIIA (Sanfillipo Syndrome) affects around 1 in 70,000 children globally and is the most common and severe variant of type 3. Children diagnosed are typically not expected to live past their late teenage years. Types B-D respectively show increases in average life expectancy based on the severity of the phenotype. A study done in the UK found pneumonia was the leading cause of death (\~50%) and that patients were very commonly prone to respiratory infection (Lavery C, et al, 2017). Other leading causes resulted from cardiac, central nervous system, and gastrointestinal issues.
Treatment complications: MPSIII currently has no approved treatment let alone cure. Many rare diseases alike are under-researched, underrepresented, and underfunded. Children living with MPS variants and other lysosomal diseases can only be treated through approved trials that often are complicated by FDA mandates. This was the case for UX111 which received mandatory alterations to the CMC (chemistry, manufacturing, controls) of their promising gene therapy treatment before further patient care could ensue (Ultragenyx, 2025). They have not been convinced of legitimate experimental trials, and some people affected by rare genetic disease argue that molecular biological research for rare cancers is prioritized first, making it hard to create progress quickly when time is of the essence.
Why I chose this project: It intrigued me when I came across a video of it by chance informing viewers of a childhood disorder that resembled that of Alzheimer's. To quote Cara O'neil, a mother of an MPS patient, "We cheer on our children for every little milestone, every gain. How does a parent respond when it's going in the other direction?". It is incredibly sad to think how these children were forming full sentences and acting as any happy child would, before the disease took that potential from them. I hope through my project I can obtain a deeper understanding of what working on the molecular biology behind cases like these is like, as well as the journey towards a cure. Hopefully as an added benefit I can spread some awareness to this cause in the process, so that families around the globe can see progress for their children. Apart from research, I have contacted the Canadian MPS society to see if there is any other ways to help.
Method
| Control | Manipulated | Responding |
|---|---|---|
| Non mutated relevant genes (SGHS, NAGLU, HGSNAT, GNS) | Changes in the sequence (mutations) | Results in MPSIII or does not |
Materials:
- NCBI Blast
- NCBI Genome Data Viewer
- EMBOSS needle pairwise sequence alignment
- OMIM
- HGMD
Procedure: 1.Use NCBI Genome viewer examine MPSIII relevant data sets 2. Acquire accession numbers and FASTA sequences for known genes: SGHS\, NAGLU\, HGSNAT\, GNS. Acquire the same for control. 3. Set database to human genome. 4. Run NCBI blastn to find matching data sets. 5. Select all matches on the same chromosome and observe graphic summary 6. Analyze e- values\, look for a relatively high value to indicate mutations. 7. Make sure the query and subject have the same name so it comes from the same locus. 8. Open the pairwise alignment to look for mutations and note the qualitative data to what kind of mutations occurred and quantitative data of how many there were. 9. Compare with known data from research to determine if it would result in MPSIII.
Research
Methods of Detecting MPS: Newborn screening: Although this method can sometimes result in false positives, it is a crucial instrument for ensuring early diagnosis in lysosomal storage disorders as it can prevent GAG build up from progressing as well as benefit patients of subtypes that do not show geno-phenotype correlations. Isobaric tags are different isotopic compositions (aka elements) with the same mass, allowing them to bind to different peptides in blood samples without other influencing factors. Different types of MPS have been tested with different sample sizes. After mixing blood samples together, mass spectrometry uses CID (collision induced dissociation) to fragment peptides by generating kinetic energy from rapid neutral gas molecule-ion collisions in order to break bonds. This releases reporter ions that allow for quantitative data about the protein's relative abundance to be measured. [14,15,16]
Treatment developments: UX111(for MPSIIA): UX111 is an experimental treatment that injects a healthy version of the SGHS gene using an AAV9 viral vector, which is a non-pathogenic virus to infiltrate the body and act as a helper cell. This specific kind (adeno-associated virus) can cross the blood-brain barrier and infect motor neurons which makes it efficient. Initial tests began in 2020 and the treatment was previously known as ABO-102. Patients who participated in the experimental trials showed an increase in Bayley-III scores, which is a developmental standardized test that indicates whether a child has properly developing neurological abilities (ex. language skills, problem solving, motor tuning, social responses, etc.). Results throughout 4 cohorts that received low-higher doses simultaneously reported less heparan sulfate concentration within cerebral spinal fluid, making it successful. However the follow-up study will soon be conducted meaning not all adverse effects are yet known, although no extremely alarming instances have been reported so far. [7, 17,]
Heparan Sulfate biochemistry summary: -is what clogs the lysosomes in MPSIII. -Glycosaminoglycan family (GAG) (fluids responsible for skin and joint health), structurally similar to heparin. -a proteoglycan (core protein with 1 or more attachment of GAG chains forming a macromolecule that supports tissue structure) -found all over cell surfaces, inside mammal tissues, & extracellular matrix. -Linear polysaccharide (repeating polymers of large carbohydrates) connected with glycosidic bonds (condensation reaction) -the enzyme for type 3A most directly breaks it down, meaning the lack of it is likely why it is the most severe subtype.
MPSIII complications/indicators: -neurological impairments (loss of speech, motor control, memory). -joint stiffness -bone deformation -difficulty breathing -hearing loss -vision clouding -cardiac complications -coarse facial hair very young
General differences of Mucopolysaccharidosis (MPS) between the four subcategories: MPSIII is an autosomal recessive disorder, meaning a child would have to receive both mutated recessive genes. However despite this there are still some differences between how each subtype works:
| Type | Genotype-phenotype correlation? | Gene | Enzyme | Chromosomal location |
|---|---|---|---|---|
| A | Yes | SGSH | heparan N-sulfatase | 17q25.3 |
| B | Yes | NAGLU | alpha-N-acetylglucosaminidase | 17q21.2 |
| C | No | HGSNAT | acetyl CoA:alpha-glucosaminide acetyltransferase | 8p11.21-p11.1 |
| D | No | GNS | N-acetylglucosamine 6-sulfatase | 12q14.3 |
MPSIIIE: MPSIIIE cases have only been reported in animal models and it's data will not be considered for this project.
Data
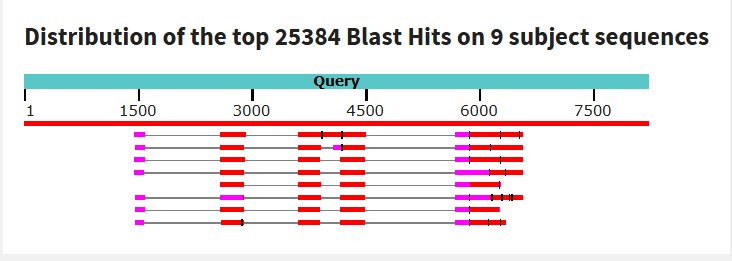
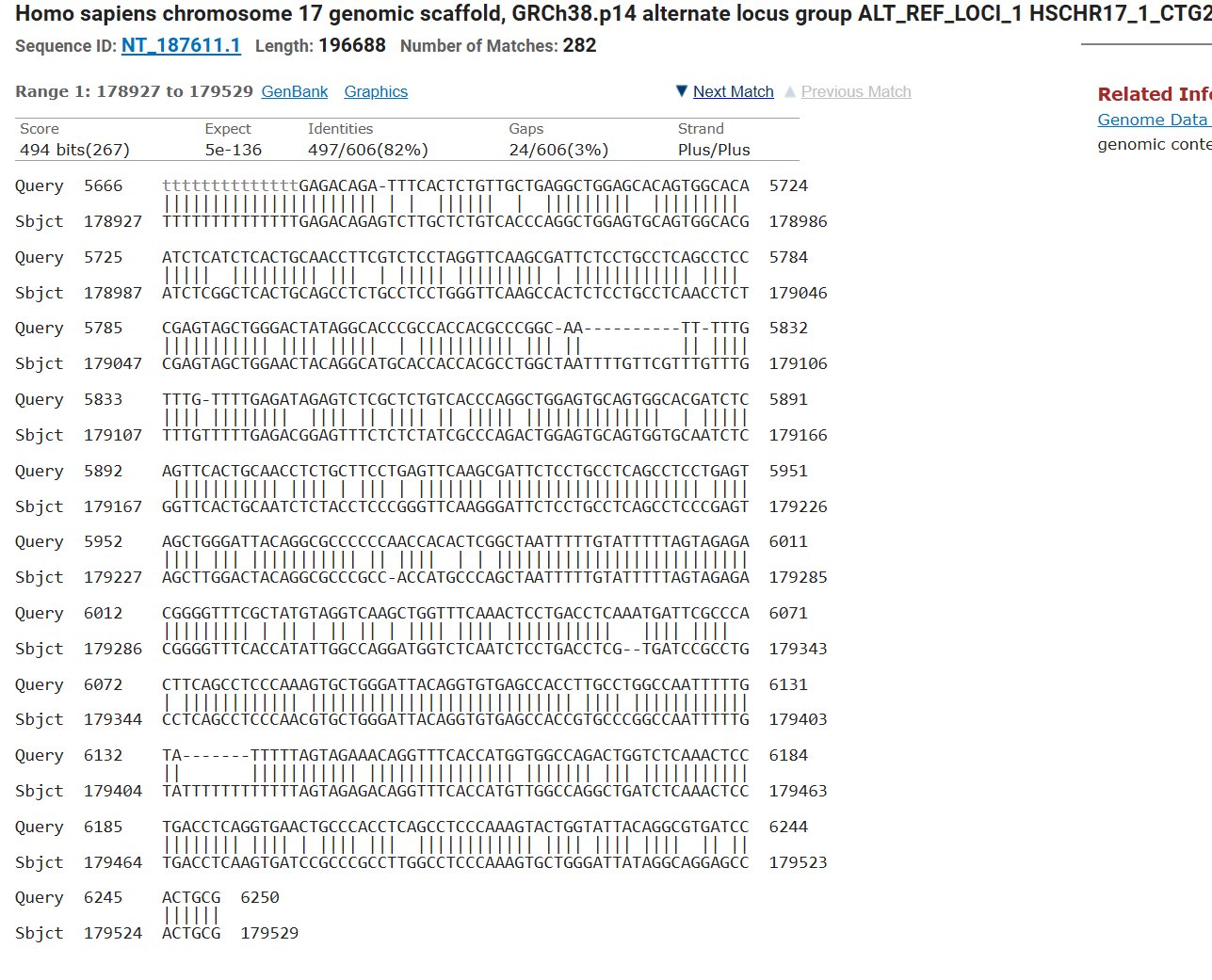
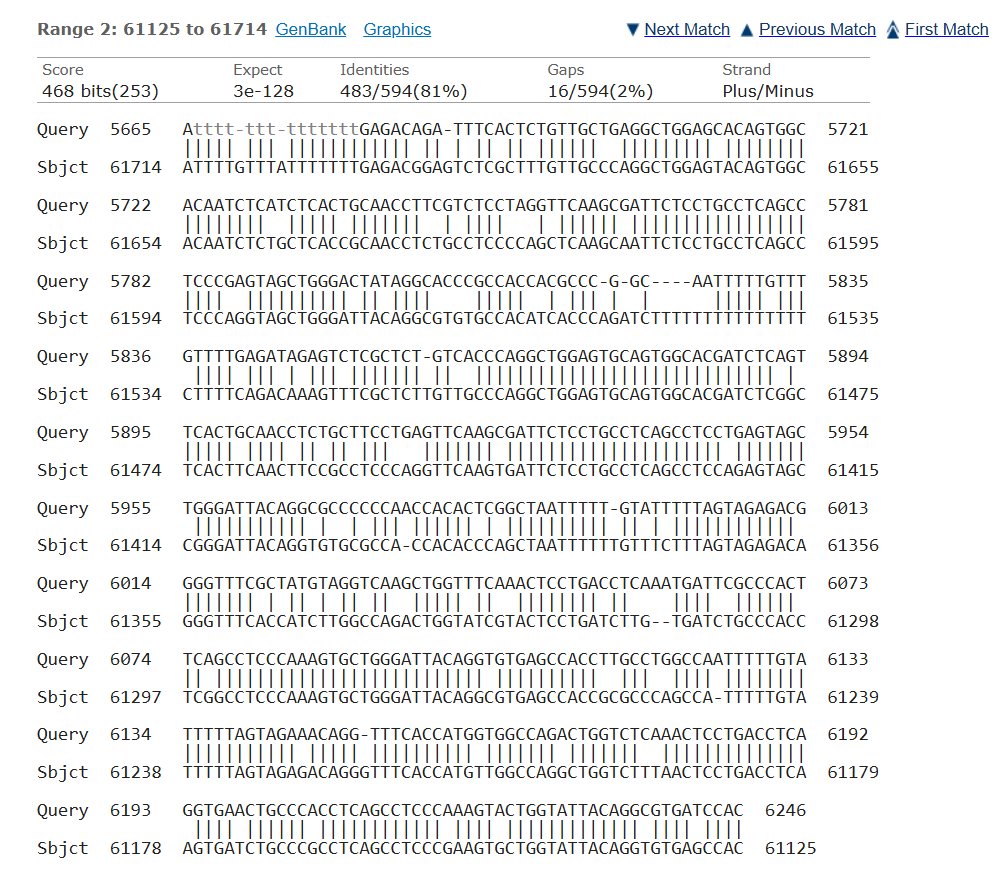

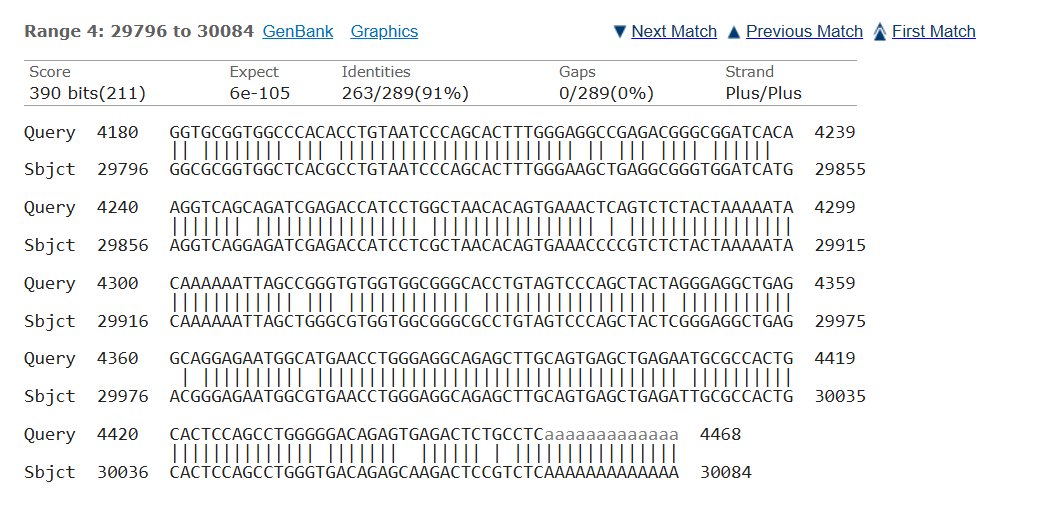
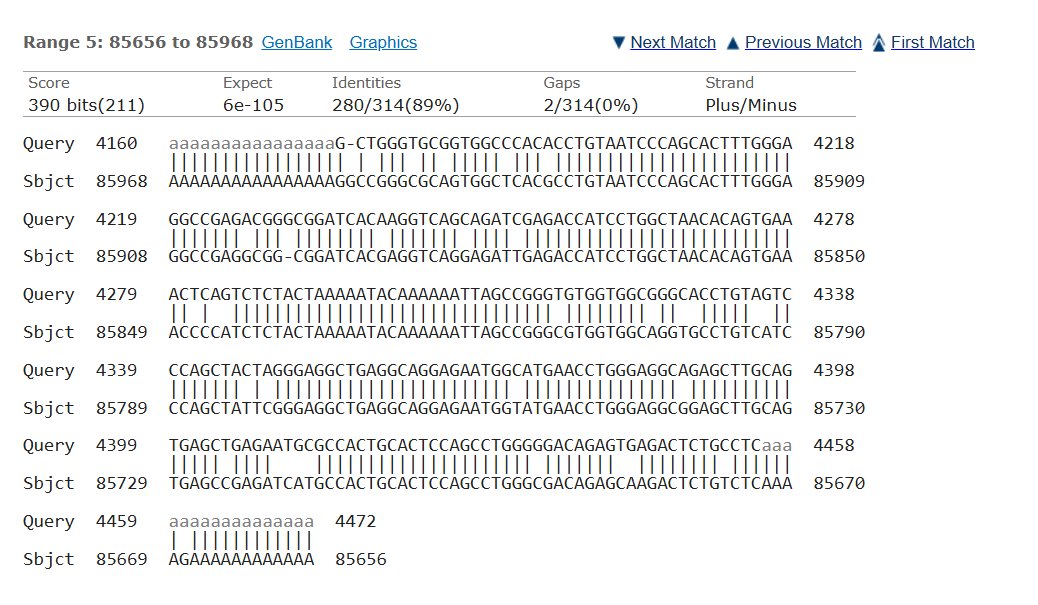
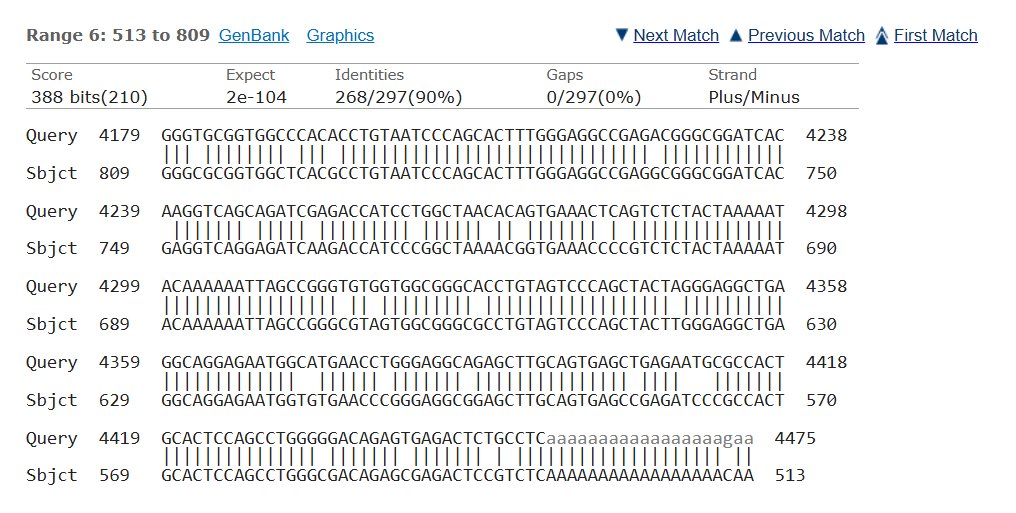
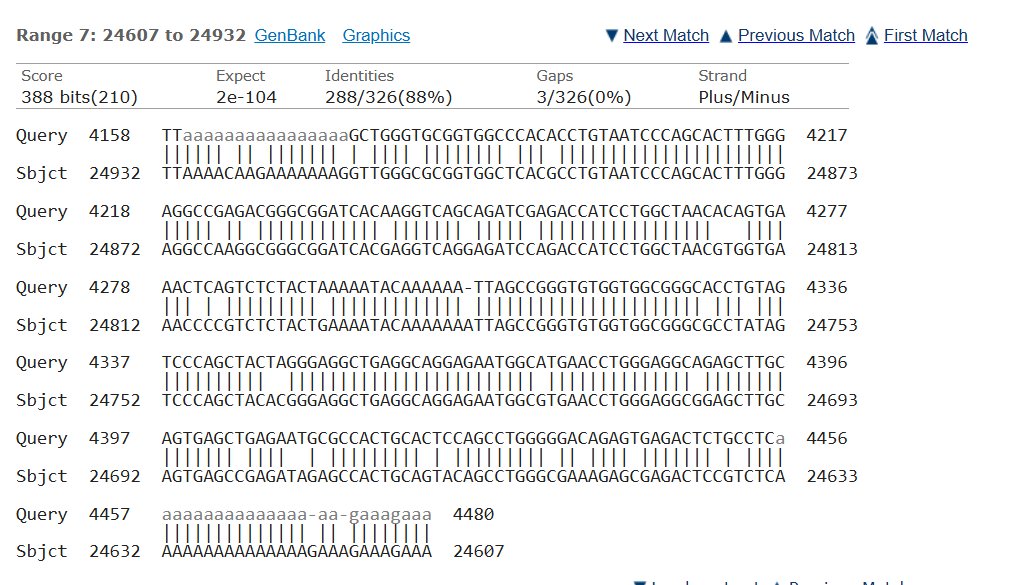
Blast was run for the NAGLU sequence and found an 82.01% match on the same chromosome with an e- value of 5e-135 making it possible that the amount of mutations in comparison to the healthy control would mean MPS would be at risk, but since they showed average trends this could also not have a huge effect.
Conclusion
The prevention and treatment of MPS is continuing to develop and genetic therapies have shown promising results. Early detection through newborn screening may also benefit a more diverse array of MPS types and subtypes in the future. As the industry continues in trials I conclude that other forms of MPS the don't show through phenotype will also be able to be treated with gene therapy and that it will be the main approach to MPSIIA once approved. In the future I want to continue my investigation into other types of MPS and genetic diseases and hopefully apply my knowledge there.
Citations
Citations:
1. Fedele\, A. (2015). Sanfilippo syndrome: causes\, consequences\, and treatments. The Application of Clinical Genetics\, 8(2015)\, 269. https://doi.org/10.2147/tacg.s57672 2. Fernández-Hernández\, L.\, Reyna-Fabián\, M. E.\, Alcántara-Ortigoza\, M. A.\, Aláez-Verson\, C.\, Flores-Lagunes\, L. L.\, Carrillo-Sánchez\, K.\, & González-del Angel\, A. (2022). Unusual Clinical Manifestations in a Mexican Patient with Sanfilippo B Syndrome. Diagnostics (2075-4418)\, 12(5)\, N.PAG–N.PAG. https://doi.org/10.3390/diagnostics12051268 3. HEMSLEY\, K.\, & HOPWOOD\, J. (2005). Development of motor deficits in a murine model of mucopolysaccharidosis type IIIA (MPS-IIIA). Behavioural Brain Research\, 158(2)\, 191–199. https://doi.org/10.1016/j.bbr.2004.08.019 4. Simon Davis\, D. A.\, & Parish\, C. R. (2013). Heparan Sulfate: A Ubiquitous Glycosaminoglycan with Multiple Roles in Immunity. Frontiers in Immunology\, 4. https://doi.org/10.3389/fimmu.2013.00470 5. Walton\, K. (2025\, July 14). FDA issues CRL for Ultragenyx’s UX111 gene therapy; company remains optimistic. Cure Sanfilippo Foundation | Accelerating Discovery of a Cure for Sanfilippo Syndrome. https://curesanfilippofoundation.org/2025/07/fda-issues-crl-for-ultragenyx-ux111-gene-therapy/ 6. Canadian MPS Society . (2022\, October 4). Types and Treatments of MPS - mpssociety. Mpssociety. https://www.mpssociety.ca/types-of-mps/ 7. Ultragenyx Pharmaceutical Inc. (2024\, October 14). UX111 for Sanfilippo syndrome type A (MPS IIIA)—Ultragenyx. Ultragenyx. https://www.ultragenyx.com/our-research/pipeline/ux111-for-mps-iiia/ 8. Belting\, M.\, Borsig\, L.\, Fuster\, M. M.\, Brown\, J. R.\, Persson\, L.\, Fransson\, L.-Å.\, & Esko\, J. D. (2002). Tumor Attenuation by Combined Heparan Sulfate and Polyamine Depletion. Proceedings of the National Academy of Sciences of the United States of America\, 99(1)\, 371–376. http://www.jstor.org/stable/3057544
9. Çelik B\, Tomatsu SC\, Tomatsu S\, Khan SA. Epidemiology of Mucopolysaccharidoses Update. Diagnostics (Basel). 2021 Feb 10;11(2):273. doi: 10.3390/diagnostics11020273. PMID: 33578874; PMCID: PMC7916572.
10. Puckett\, Y.\, Mallorga-Hernández\, A.\, & Montaño\, A. M. (2021). Epidemiology of mucopolysaccharidoses (MPS) in United States: challenges and opportunities. Orphanet Journal of Rare Diseases, 16(1). https://doi.org/10.1186/s13023-021-01880-8
11. Prognosis of Sanfilippo*. (n.d.). Cure Sanfilippo Foundation | Accelerating Discovery of a Cure for Sanfilippo Syndrome. https://curesanfilippofoundation.org/what-is-sanfilippo/for-physicians/prognosis/
12. Lavery\, C.\, Hendriksz\, C. J.\, & Jones\, S. A. (2017). Mortality in patients with Sanfilippo syndrome. Orphanet journal of rare diseases, 12(1), 168. https://doi.org/10.1186/s13023-017-0717-y 13. Rauf\, A.\, Badoni\, H.\, Abu-Izneid\, T.\, Olatunde\, A.\, Rahman\, Md. M.\, Painuli\, S.\, Semwal\, P.\, Wilairatana\, P.\, & Mubarak\, M. S. (2022). Neuroinflammatory Markers: Key Indicators in the Pathology of Neurodegenerative Diseases. Molecules\, 27(10)\, 3194. https://doi.org/10.3390/molecules27103194
14. Chien\, Y. H.\, Lee\, N. C.\, Chen\, P. W.\, Yeh\, H. Y.\, Gelb\, M. H.\, Chiu\, P. C.\, Chu\, S. Y.\, Lee\, C. H.\, Lee\, A. R.\, & Hwu\, W. L. (2020). Newborn screening for Morquio disease and other lysosomal storage diseases: results from the 8-plex assay for 70\,000 newborns. Orphanet journal of rare diseases, 15(1), 38. https://doi.org/10.1186/s13023-020-1322-z
15. Ding\, S.\, & Han\, L. (2022). Newborn screening for genetic disorders: Current status and prospects for the future. Pediatric investigation, 6(4), 291–298. https://doi.org/10.1002/ped4.12343
16. Pottiez\, G.\, Wiederin\, J.\, Fox\, H. S.\, & Ciborowski\, P. (2012). Comparison of 4-plex to 8-plex iTRAQ quantitative measurements of proteins in human plasma samples. Journal of proteome research, 11(7), 3774–3781. https://doi.org/10.1021/pr300414z
17. (2025). Clinicaltrials.gov. https://clinicaltrials.gov/study/NCT02716246?term=abo-102&viewType=Table&rank=3
Acknowledgement
-Some use of Microsoft Copilot Ai was used to help navigate software and clear up small questions/definitions but did not contribute to the project content otherwise. -I'd like to acknowledge and thank Akshita Rawat and Sara Waqas for offering guidance on how to format this project.