Combating Antifungal Resistance Through Secondary “Rescue” Mutations in Candida auris
Zafina Zaman
STEM Innovation Academy High School
Grade 11
Presentation
No video provided
Problem
Antimicrobial resistance (AMR) is recognized by the World Health Organization (WHO) as one of the top global public health threats of the 21st century. Drug-resistant infections are projected to cause millions of deaths annually if left unaddressed, placing increasing strain on healthcare systems worldwide. While AMR is often associated with bacteria, fungal resistance is an equally urgent and rapidly emerging concern. Fungal infections are particularly concerning because the number of available antifungal drug classes is limited compared to antibacterial agents. As resistance emerges, treatment options rapidly narrow, increasing the urgency for innovative approaches that extend the effectiveness of existing therapies. Preserving current drugs is crucial, and making new drugs is highly costly and time-intensive. Widespread antifungal use in hospitals and immunocompromised populations creates strong selective pressure, accelerating the emergence of resistance-conferring mutations.
Among resistant fungal pathogens, Candida auris has been designated by the WHO as one of the 19 priority fungal pathogens and is described as a “serious global health threat.” Since its identification, C. auris has been detected in approximately 40 countries and is associated with high mortality rates, with invasive infections resulting in death in roughly one-third of cases. A major contributor to this threat is resistance to fluconazole, a frontline antifungal medication widely used to treat Candida infections. Fluconazole functions by binding to lanosterol 14α-demethylase, an essential fungal enzyme involved in ergosterol biosynthesis. However, mutations such as Y132F alter the structure of this enzyme and reduce drug effectiveness. Current approaches to antifungal resistance largely focus on designing new drugs or modifying existing compounds to improve binding interactions. While many studies quantify changes in binding affinity, fewer investigate how resistance mutations reshape the three-dimensional architecture of the binding pocket and alter ligand orientation.
Therefore, alternative structure-guided strategies are needed to better understand how resistance mutations alter protein geometry and drug binding. By examining how these mutations reshape the binding pocket and influence ligand orientation, it may be possible to identify compensatory mechanisms that restore drug susceptibility without relying solely on the development of entirely new drugs. Computational modelling provides a cost-effective approach to analyzing the structural consequences of resistance mutations before experimental validation, enabling the targeted identification of promising structural interventions. In this project, I investigate the molecular geometry underlying antifungal resistance using established computational docking and structural visualization tools. By shifting the focus from interaction strength to binding pocket architecture and flexibility, this research offers a structural perspective on how resistance emerges and how it may be countered.
Method
Materials:
- Device with internet access
- PyMOL
- ColabFold
- CB-Dock2
- PLIP
- Ramplot
- UniProt
All computational analyses were performed using default parameters unless otherwise specified to ensure reproducibility. All structural predictions and docking simulations were conducted independently for each variant under consistent computational conditions. Structural overlays and interaction analyses were performed using standardized visualization settings to minimize analytical bias. This ensured reproducibility and objective comparative assessment across all tested mutations.
Baseline Sequence Acquisition and Structural Modelling
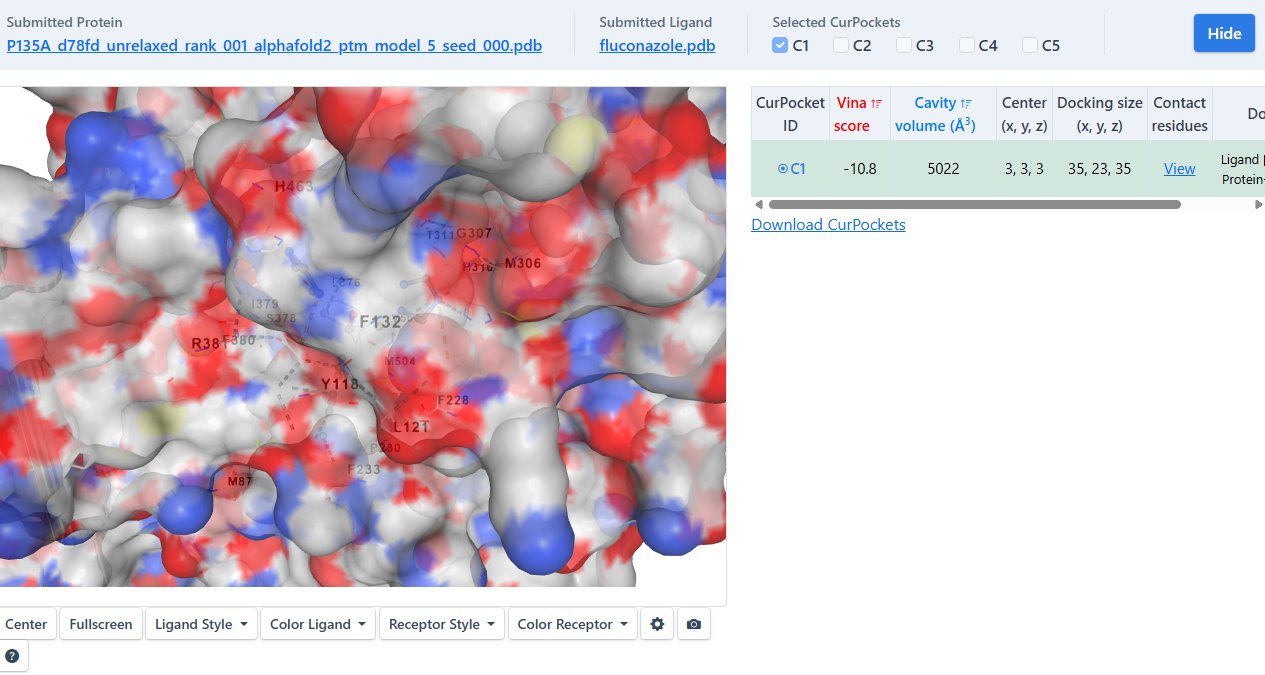
The amino acid sequence of Candida auris ERG11 (lanosterol 14α-demethylase) wild-type protein was obtained from the NCBI Protein Database. The sequence was cross-referenced against UniProt to confirm its accuracy. The primary amino acid sequence was submitted to ColabFold to generate a predicted three-dimensional protein structure using AlphaFold2-based modelling. The highest-confidence structural prediction (based on pLDDT confidence scores) was selected for downstream analysis. The predicted structure was downloaded as a PDB file and visually inspected in PyMOL to confirm structural integrity and absence of major folding anomalies Wild-Type Docking and Binding Characterization The wild-type ERG11 structure was uploaded to CB-Dock2 for molecular docking with fluconazole. CB-Dock2 automatically identified potential binding cavities and generated docking poses ranked by binding affinity (kcal/mol). The top-ranked docking pose, based on the lowest binding energy and biologically plausible orientation within the active site, was selected for analysis. Binding affinity values were recorded. The docked protein–ligand complex was downloaded and imported into PyMOL for visualization and structural inspection. The complex was then submitted to PLIP to quantify:
- Hydrogen bonds
- Hydrophobic interactions
- π–π stacking interactions
- Overall contact number
These interaction metrics established the baseline binding profile of the wild-type enzyme.
Modelling the Y132F Resistance Mutation
To simulate antifungal resistance, the Y132F mutation — the most common mutation in Candida auris — was introduced into the ERG11 amino acid sequence by manually substituting tyrosine (Y) at position 132 with phenylalanine (F). The mutated sequence was submitted to ColabFold to generate a new predicted three-dimensional structure. Structural confidence scores were evaluated to ensure proper folding. The Y132F structure was then docked with fluconazole using the same CB-Dock2 parameters applied to the wild-type model to maintain experimental consistency. Binding affinity values were recorded and compared to wild-type values.
The docked Y132F complex was analyzed using:
- PyMOL for structural overlay comparison with wild-type
- PLIP for interaction quantification
Comparisons focused on:
- Loss or alteration of hydrogen bonds
- Changes in ligand orientation
- Alterations in binding pocket geometry
- Reduction in the overall contact number
This established the structural and energetic consequences of the resistance mutation.
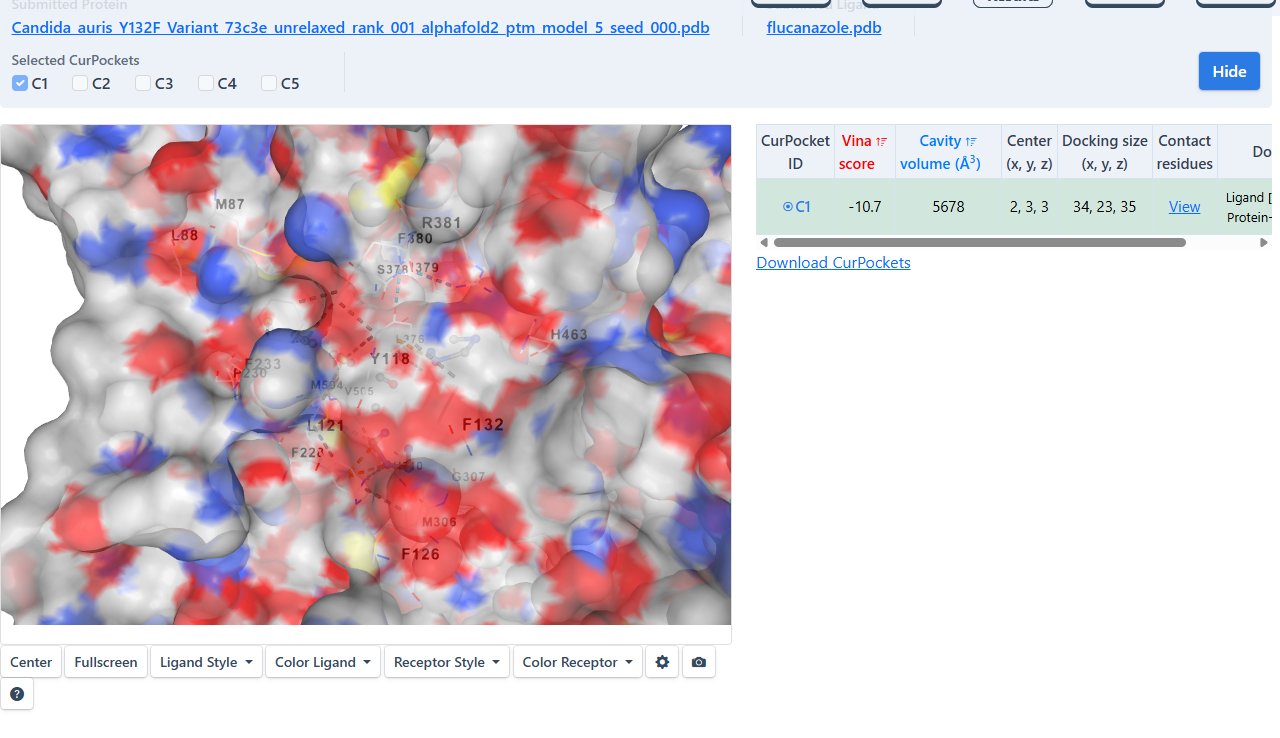 To ensure valid comparative analysis, identical docking parameters were maintained across all simulations. The same predicted binding cavity identified by CB-Dock2 was used for all variants, and default exhaustiveness and scoring functions were preserved. This controlled approach ensured that differences in binding affinity and ligand orientation were attributable to structural mutations rather than variations in docking configuration.
To ensure valid comparative analysis, identical docking parameters were maintained across all simulations. The same predicted binding cavity identified by CB-Dock2 was used for all variants, and default exhaustiveness and scoring functions were preserved. This controlled approach ensured that differences in binding affinity and ligand orientation were attributable to structural mutations rather than variations in docking configuration.
Identification of Candidate Residues for Secondary Mutation
Residues within close spatial proximity to the fluconazole binding pocket (≤5 Å) were identified using PyMOL distance measurements. Candidate residues for secondary mutation were selected based on:
- Side-chain polarity
- Hydrogen bonding potential
- Steric bulk
- Orientation relative to the altered Y132F residue
- Potential to restore binding pocket geometry
Only residues plausibly capable of compensating for structural disruption were considered. This ensured mutations were hypothesis-driven rather than random.
The table below was created and used to understand the role of each amino acid:
| Letter | Amino Acid Name | Properties |
|---|---|---|
| A | Alanine | Small, non polar |
| R | Arginine | Positively charged (basic) |
| N | Asparagine | Polar, uncharged |
| D | Aspartic acid | Negatively charged (acidic) |
| C | Cysteine | Polar. Can form disulphide bonds |
| E | Glutamic acid | Negatively charged (acidic) |
| Q | Glutamine | Polar, uncharged |
| G | Glycine | Very small, flexible |
| H | Histidine | Positive/neutral (can switch), often catalytic, aromatic |
| I | Isoleucine | Hydrophobic |
| L | Leucine | Hydrophobic |
| K | Lysine | Positively charged (basic) |
| M | Methionine | Hydrophobic, sulfur-containing |
| F | Phenylalanine | Aromatic, hydrophobic |
| P | Proline | Rigid, causing kinks in the protein chain |
| S | Serine | Polar, can form H-bonds |
| T | Threonine | Polar, can form H-bonds |
| W | Tryptophan | Large aromatic, hydrophobic |
| Y | Tyrosine | Aromatic, polar OH group |
| V | Valine | Hydrophobic |
Iterative In-Silico Secondary Mutation Screening
An iterative computational mutagenesis pipeline was developed to evaluate compensatory structural variants. For each candidate secondary mutation:
- The amino acid substitution was introduced into the Y132F sequence.
- The modified sequence was submitted to ColabFold for 3D structure prediction.
- The predicted structure was docked with fluconazole using CB-Dock2.
- Binding affinity values were recorded.
- Protein–ligand interactions were quantified using PLIP.
- Ligand orientation and binding pocket geometry were evaluated in PyMOL.
Each variant was evaluated against predefined criteria: A mutation was considered potentially compensatory if it:
- Improved binding affinity relative to Y132F
- Increased hydrogen bonding or favourable interactions
- Restored ligand orientation toward wild-type geometry
- Maintained structural stability
This process was repeated systematically across multiple candidate residues.
A variant was classified as compensatory only if it demonstrated measurable improvement in geometric alignment toward the wild-type conformation while maintaining equal or improved binding affinity relative to the Y132F variant. Variants that increased interaction count without restoring spatial orientation were not considered successful.
A total of 16 secondary-mutation variants were systematically evaluated before identifying structurally significant candidates. Each variant underwent identical modelling, docking, and interaction analysis procedures. This iterative screening approach ensured that final candidates emerged from structured comparative evaluation rather than isolated observation.
Structural Stability Validation
For the most promising compensatory variant(s), structural stability was assessed using Ramachandran plots generated with Ramplot. The distribution of phi (φ) and psi (ψ) backbone angles was examined to ensure:
- The majority of residues fell within favoured regions
- No significant increase in disallowed conformations relative to wild-type
This ensured that improved binding affinity was achieved without compromising overall protein stability. Binding affinity values, interaction counts, and geometric features were compiled for comparative analysis.
Research
Emergence and Resistance of Candida auris
Candida auris is an emerging multidrug-resistant fungal pathogen first isolated in 2007 from the external ear canal of a patient in Japan. Since its identification, it has rapidly spread across South Asia, Africa, South America, and other regions, establishing itself as a significant global health threat. Unlike many Candida species that primarily colonize the gastrointestinal tract, C. auris preferentially colonizes the skin, facilitating transmission in healthcare environments. Antifungal resistance in C. auris is acquired rather than intrinsic. Distinct global clades have independently developed resistance to azole antifungals, demonstrating significant genetic diversity. Minimum inhibitory concentration (MIC) values for fluconazole have been observed to increase dramatically in resistant isolates, with reported ranges escalating from 16–64 µg/mL to as high as 64–256 µg/mL in certain populations. Because separate clades have independently evolved azole resistance, MIC values vary geographically, highlighting the evolutionary plasticity of this organism. Prior antifungal exposure is strongly associated with rapid acquisition of resistance. The most common mechanisms of antifungal resistance include:
- Mutations in the antifungal target that reduce drug binding
- Overexpression of the target enzyme through transcriptional or ploidy alterations
- Overexpression of efflux pumps that actively remove antifungal agents from the cell
Among these mechanisms, mutations in the azole target enzyme ERG11 are among the most clinically significant contributors to fluconazole resistance.
Mechanism of Fluconazole and ERG11-Mediated Resistance
Fluconazole is a triazole antifungal that inhibits lanosterol 14α-demethylase, an essential enzyme in the ergosterol biosynthesis pathway. This enzyme is encoded by the ERG11 gene. Ergosterol is a critical structural component of fungal cell membranes; inhibition of ERG11 disrupts membrane integrity and impairs fungal growth. Resistance frequently arises through mutations in ERG11 that alter the structure of the drug-binding pocket. One clinically prevalent mutation in C. auris is Y132F, in which tyrosine is replaced by phenylalanine. This substitution removes a hydroxyl group capable of hydrogen bonding while retaining an aromatic ring structure. As a result, subtle but significant alterations in local pocket geometry and polarity may occur. More broadly, drug resistance mutations often occur in regions that directly interact with therapeutic inhibitors. These mutations can:
- Introduce steric clashes
- Remove favourable hydrogen bonds
- Alter electrostatic interactions
- Destabilize conformations preferred by the inhibitor
In oncology, similar mechanisms are observed in kinase-driven leukemias, where mutations in ATP-binding sites or activation loops prevent effective inhibitor binding by reshaping the binding pocket. These examples illustrate that resistance frequently arises from structural remodelling rather than complete loss of enzymatic function.
Structural Determinants of Protein–Ligand Binding
Protein–ligand interactions depend on multiple structural factors, including hydrogen bonding, hydrophobic packing, electrostatic complementarity, and three-dimensional shape compatibility. While binding affinity is often quantified in terms of interaction energy or contact number, optimal binding requires proper geometric alignment between ligand and binding pocket. Even small conformational shifts in side-chain orientation or backbone flexibility can significantly influence ligand positioning. The geometry of a binding pocket is shaped not only by direct contact residues but also by neighbouring amino acids that influence local structural stability and flexibility. Proline plays a unique structural role in proteins due to its rigid cyclic side chain, which restricts backbone rotation and constrains conformational flexibility. In contrast, smaller residues such as alanine introduce minimal steric hindrance and allow greater rotational freedom within the peptide backbone. Thus, local residue composition can strongly influence binding pocket adaptability. These principles suggest that resistance mutations may disrupt not only interaction strength but also spatial alignment and conformational dynamics within the binding site.
Secondary (Suppressor) Mutations as Structural Compensators
Drug resistance mutations are often interpreted as direct modifications of drug-binding residues. However, increasing evidence suggests that resistance frequently arises from broader structural consequences rather than isolated contact loss. Primary resistance mutations can introduce subtle distortions in protein folding, alter backbone strain, or destabilize conformational states necessary for optimal ligand accommodation. A well-documented example of this phenomenon was demonstrated in studies of TEM-1 β-lactamase, where a resistance-associated mutation caused structural instability and increased misfolding. A secondary mutation was later identified that suppressed this instability. Importantly, the suppressor mutation did not directly restore drug-binding chemistry. Instead, it improved global protein stability, reduced aggregation, and increased the amount of properly folded enzyme. When combined with the resistance mutation, enzymatic function was restored. This finding established a critical principle: Secondary mutations can compensate for the structural consequences of resistance mutations without directly reversing the original biochemical interaction changes. Rather than “undoing” the resistance mutation, suppressor mutations often:
- Rebalance local structural strain
- Restore conformational stability
- Improve folding efficiency
- Reestablish favourable geometric architecture
In many systems, resistance mutations impose energetic or conformational penalties. Compensatory mutations can offset these penalties by stabilizing adjacent regions or redistributing backbone flexibility. This concept is fundamental to antifungal resistance. If a mutation such as Y132F alters binding pocket geometry or rigidity, the loss of susceptibility may reflect structural distortion rather than purely the loss of a hydrogen bond. A secondary mutation could therefore restore drug accommodation by correcting architectural imbalance rather than increasing interaction count. This theoretical framework shifts the goal from “adding interactions” to “restoring structural harmony.”
Conceptual Framework: Geometry and Flexibility in Resistance Reversal
Protein–ligand binding is not determined solely by the number of hydrogen bonds or contact residues. It depends critically on three-dimensional complementarity — the spatial fit between ligand and binding pocket. Binding affinity emerges from:
- Shape complementarity
- Optimal orientation
- Electrostatic alignment
-
Dynamic adaptability of the pocket
A resistance mutation may preserve many interactions yet subtly alter ligand orientation, depth of insertion, or rotational positioning. Even small angular deviations can reduce effective binding despite similar contact counts.
This highlights the importance of structural geometry. Additionally, proteins are not rigid structures. Binding pockets exist within dynamic frameworks. Local backbone flexibility allows residues to adjust in response to ligand presence. If flexibility is reduced — for example, through rigid residues such as proline — the pocket may become less adaptable and less capable of accommodating structural disturbances caused by mutation. Proline is uniquely restrictive because its cyclic side chain locks the backbone phi angle, limiting conformational freedom. This rigidity can stabilize local structure, but it can also reduce adaptive flexibility in regions requiring subtle geometric adjustments. In contrast, alanine possesses a minimal methyl side chain. It introduces minimal steric bulk and allows significantly greater backbone rotational freedom. Because of this, alanine is widely used in structural biology as a neutral substitution to probe functional and structural contributions of specific residues. The study “Rapid Mapping of Protein Functional Epitopes by Combinatorial Alanine Scanning” demonstrated that alanine substitution can systematically evaluate the functional contribution of residues without introducing dramatic structural perturbation. Alanine scanning works because alanine removes side-chain complexity while largely preserving backbone structure. This allows researchers to determine whether side-chain bulk, polarity, or rigidity contributes critically to protein function. The theoretical implication is powerful: Alanine substitutions often reduce steric interference and relieve local strain while maintaining overall fold integrity. Applying this to resistance: If Y132F introduces geometric distortion within the binding pocket, and if nearby residues contribute to pocket rigidity, replacing a rigid residue such as proline with alanine may:
- Increase backbone flexibility
- Reduce steric tension
- Allow the pocket to reconfigure dynamically
- Enable the ligand to adopt a more favourable orientation
Rather than forcing the ligand into position through steric bulk (as seen with bulky substitutions), increasing flexibility permits the protein to adaptively reshape around the ligand. This represents a fundamentally different compensatory strategy:
- Steric forcing strategy → Constrain ligand into alignment
- Flexibility restoration strategy → Allow ligand to self-align through adaptive geometry
The second approach aligns directly with principles observed in suppressor mutation literature, where restoring conformational balance is often more effective than increasing interaction density.
Understanding the Differences Between Y132F and the WT (Wild Type)
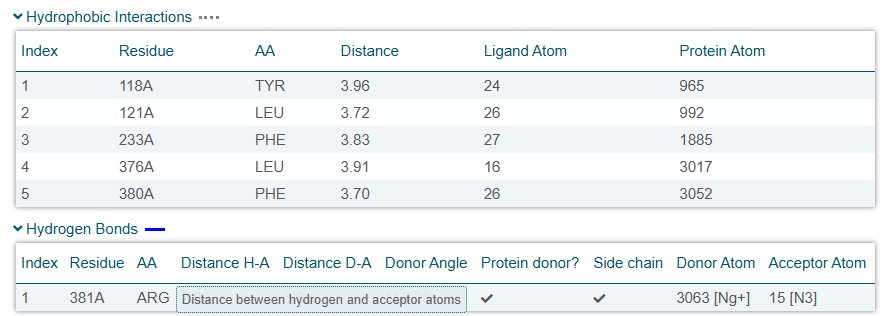
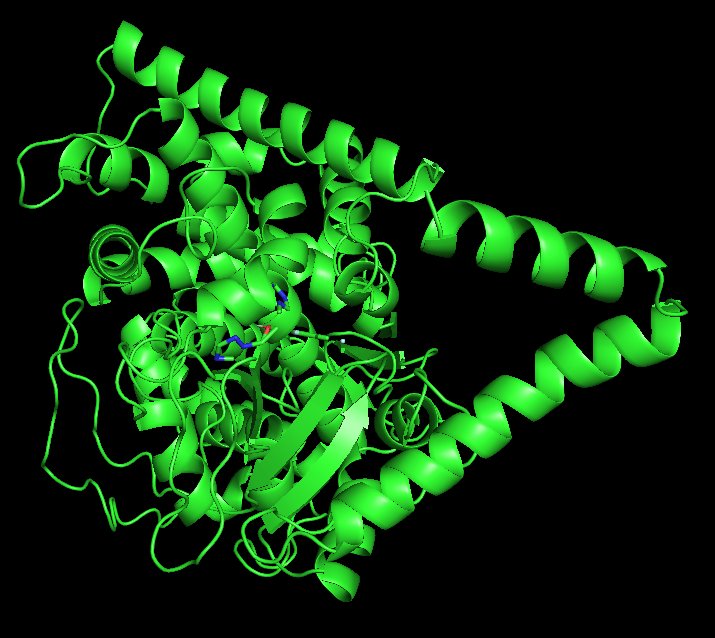
Following molecular docking analysis, the wild-type ERG11–fluconazole complex demonstrated a binding affinity of –10.7 kcal/mol, indicating strong thermodynamic favorability. In molecular docking, more negative binding affinity values correspond to stronger predicted interactions, as they reflect lower free energy states of the protein–ligand complex. In addition to favourable binding energy, PLIP analysis revealed a greater number of stabilizing contacts in the wild-type complex, including hydrogen bonds and hydrophobic interactions. These interactions contribute to ligand stabilization within the active site and help maintain optimal orientation of fluconazole relative to catalytic residues. Together, the strong binding energy and high interaction count suggest that fluconazole is structurally well accommodated within the wild-type binding pocket. In contrast, docking of the Y132F variant resulted in a reduced binding affinity and a decrease in stabilizing interactions. Although the substitution from tyrosine to phenylalanine is considered a conservative mutation (both are aromatic residues), the loss of the hydroxyl (-OH) group in phenylalanine eliminates a potential hydrogen bonding donor. This seemingly small chemical alteration significantly impacts local interaction networks within the binding pocket.
Observed Ligand Reorientation in Y132F
Structural visualization in PyMOL revealed that fluconazole adopted a rotated orientation within the Y132F binding pocket compared to the wild-type complex. In the wild-type structure, the hydroxyl group of Y132 contributes to stabilizing interactions that help anchor fluconazole in a specific orientation. When this hydroxyl group is removed in the Y132F mutation, the local hydrogen bonding network is disrupted. As a result, fluconazole shifts position and rotates within the binding cavity. This rotational displacement alters:
- The alignment of the triazole rings
- The positioning of halogen substituents
- The proximity to key catalytic residues
- The overall geometry of the binding pocket
Even subtle changes in ligand orientation can reduce binding efficiency because optimal molecular recognition depends not only on interaction strength but also on precise spatial alignment. Overlay analysis between wild-type and Y132F complexes demonstrated that resistance is not solely due to weaker interaction strength, but rather to a restructuring of binding pocket geometry that modifies ligand positioning.
Discovery of the P135W Compensatory Variant
During iterative secondary mutation screening, the P135W substitution produced an unexpected structural outcome. Upon docking analysis, the fluconazole molecule adopted an orientation that was approximately 180° rotated relative to the configuration observed in the Y132F mutant. At first glance, the binding affinity alone did not fully explain the significance of this change. The P135W mutation actually introduced a halogen bond, which is supposed to be fairly strong, but the binding affinity still stayed at -10.7, like the Y132F mutation. Structural overlay in PyMOL revealed that the rotation repositioned fluconazole into a geometry more closely resembling the wild-type alignment within the active site. This observation marked a critical shift in analytical focus. Proline (P135) is a rigid, cyclic amino acid that restricts backbone flexibility due to its unique ring structure. Substituting proline with tryptophan (W), a bulky aromatic residue, introduced:
- Increased steric volume
- Expanded π-electron surface area
- Greater potential for aromatic stacking interactions
- Altered local backbone flexibility
The presence of tryptophan at position 135 reshaped the spatial constraints of the binding pocket. Rather than merely increasing the number of contacts, the mutation altered the three-dimensional contour of the cavity itself. This steric remodelling forced fluconazole to reorient within the active site, effectively flipping its binding configuration. Importantly, this reorientation restored key spatial relationships between the ligand and catalytically relevant residues that had been disrupted in the Y132F mutation.
Data
Protein Data Table:
| PROTEIN | BINDING AFFINITY | INTERACTIONS | CODE |
|---|---|---|---|
| ERG11 Wild Type | -10.8 | Hydrophobic→ 3
|
MALKDCIVDVVDRFSALPVPVKLAVLILVPIVYNLVWQFVYSLRKDRAPLVFHWVPWVGSAVVYGMQPYQFFESCREKYGDVFAFVMLGKVMTVYLGPKGHEFVLNAKLADVSAEAAYSHLTTPVFGKGVIYDCPNSRLMEQKKFAKTALTKEAFQRYVPRIQEEVLDYFKACSQFRMNERNNGVANVMKTQPEMTILTASKSLMGDDMRARFDASFAKLYSDLDKGFTPINFVFPHLPLPAYWKRDAAQQKISATYMSLINERRKTGDIVPDRDLIDSLMTNSTYKDGVKMTDQEVANLLIGVLMGGQHTSASTSAWFLLHLAEQPKLQEELYSEVLSVLADKGGSLKDLAYDDLQKMPLINQTIKETLRLHMPLHSIFRKVMNPLVVPNTKYVVPKGHYVMVSPGYAQTNEKWFPRANEFDPHRWDEETSSNIDTDAVDYGFGKVTKGVSSPYLPFGGGRHRCIGEQFAYVQLGTILATYVYNIKWRFKKDGSLPPVDYQSMVTLPMEPAEIEWEKRETCVY |
| Y132F | -10.7 | Hydrophobic→ 4
|
MALKDCIVDVVDRFSALPVPVKLAVLILVPIVYNLVWQFVYSLRKDRAPLVFHWVPWVGSAVVYGMQPYQFFESCREKYGDVFAFVMLGKVMTVYLGPKGHEFVLNAKLADVSAEAAYSHLTTPVFGKGVIFDCPNSRLMEQKKFAKTALTKEAFQRYVPRIQEEVLDYFKACSQFRMNERNNGVANVMKTQPEMTILTASKSLMGDDMRARFDASFAKLYSDLDKGFTPINFVFPHLPLPAYWKRDAAQQKISATYMSLINERRKTGDIVPDRDLIDSLMTNSTYKDGVKMTDQEVANLLIGVLMGGQHTSASTSAWFLLHLAEQPKLQEELYSEVLSVLADKGGSLKDLAYDDLQKMPLINQTIKETLRLHMPLHSIFRKVMNPLVVPNTKYVVPKGHYVMVSPGYAQTNEKWFPRANEFDPHRWDEETSSNIDTDAVDYGFGKVTKGVSSPYLPFGGGRHRCIGEQFAYVQLGTILATYVYNIKWRFKKDGSLPPVDYQSMVTLPMEPAEIEWEKRETCVY |
| I131F | -10.5 | -- | MALKDCIVDVVDRFSALPVPVKLAVLILVPIVYNLVWQFVYSLRKDRAPLVFHWVPWVGSAVVYGMQPYQFFESCREKYGDVFAFVMLGKVMTVYLGPKGHEFVLNAKLADVSAEAAYSHLTTPVFGKGVYFDCPNSRLMEQKKFAKTALTKEAFQRYVPRIQEEVLDYFKACSQFRMNERNNGVANVMKTQPEMTILTASKSLMGDDMRARFDASFAKLYSDLDKGFTPINFVFPHLPLPAYWKRDAAQQKISATYMSLINERRKTGDIVPDRDLIDSLMTNSTYKDGVKMTDQEVANLLIGVLMGGQHTSASTSAWFLLHLAEQPKLQEELYSEVLSVLADKGGSLKDLAYDDLQKMPLINQTIKETLRLHMPLHSIFRKVMNPLVVPNTKYVVPKGHYVMVSPGYAQTNEKWFPRANEFDPHRWDEETSSNIDTDAVDYGFGKVTKGVSSPYLPFGGGRHRCIGEQFAYVQLGTILATYVYNIKWRFKKDGSLPPVDYQSMVTLPMEPAEIEWEKRETCVY |
| N132C | -10.2 | -- | MALKDCIVDVVDRFSALPVPVKLAVLILVPIVYNLVWQFVYSLRKDRAPLVFHWVPWVGSAVVYGMQPYQFFESCREKYGDVFAFVMLGKVMTVYLGPKGHEFVLNAKLADVSAEAAYSHLTTPVFGKGVIFDCPCSRLMEQKKFAKTALTKEAFQRYVPRIQEEVLDYFKACSQFRMNERNNGVANVMKTQPEMTILTASKSLMGDDMRARFDASFAKLYSDLDKGFTPINFVFPHLPLPAYWKRDAAQQKISATYMSLINERRKTGDIVPDRDLIDSLMTNSTYKDGVKMTDQEVANLLIGVLMGGQHTSASTSAWFLLHLAEQPKLQEELYSEVLSVLADKGGSLKDLAYDDLQKMPLINQTIKETLRLHMPLHSIFRKVMNPLVVPNTKYVVPKGHYVMVSPGYAQTNEKWFPRANEFDPHRWDEETSSNIDTDAVDYGFGKVTKGVSSPYLPFGGGRHRCIGEQFAYVQLGTILATYVYNIKWRFKKDGSLPPVDYQSMVTLPMEPAEIEWEKRETCVY |
| Q142S | -10.3 | -- | MALKDCIVDVVDRFSALPVPVKLAVLILVPIVYNLVWQFVYSLRKDRAPLVFHWVPWVGSAVVYGMQPYQFFESCREKYGDVFAFVMLGKVMTVYLGPKGHEFVLNAKLADVSAEAAYSHLTTPVFGKGVIFDCPNSRLMESKKFAKTALTKEAFQRYVPRIQEEVLDYFKACSQFRMNERNNGVANVMKTQPEMTILTASKSLMGDDMRARFDASFAKLYSDLDKGFTPINFVFPHLPLPAYWKRDAAQQKISATYMSLINERRKTGDIVPDRDLIDSLMTNSTYKDGVKMTDQEVANLLIGVLMGGQHTSASTSAWFLLHLAEQPKLQEELYSEVLSVLADKGGSLKDLAYDDLQKMPLINQTIKETLRLHMPLHSIFRKVMNPLVVPNTKYVVPKGHYVMVSPGYAQTNEKWFPRANEFDPHRWDEETSSNIDTDAVDYGFGKVTKGVSSPYLPFGGGRHRCIGEQFAYVQLGTILATYVYNIKWRFKKDGSLPPVDYQSMVTLPMEPAEIEWEKRETCVY |
| Q142T | -10.4 | -- | MALKDCIVDVVDRFSALPVPVKLAVLILVPIVYNLVWQFVYSLRKDRAPLVFHWVPWVGSAVVYGMQPYQFFESCREKYGDVFAFVMLGKVMTVYLGPKGHEFVLNAKLADVSAEAAYSHLTTPVFGKGVIFDCPNSRLMETKKFAKTALTKEAFQRYVPRIQEEVLDYFKACSQFRMNERNNGVANVMKTQPEMTILTASKSLMGDDMRARFDASFAKLYSDLDKGFTPINFVFPHLPLPAYWKRDAAQQKISATYMSLINERRKTGDIVPDRDLIDSLMTNSTYKDGVKMTDQEVANLLIGVLMGGQHTSASTSAWFLLHLAEQPKLQEELYSEVLSVLADKGGSLKDLAYDDLQKMPLINQTIKETLRLHMPLHSIFRKVMNPLVVPNTKYVVPKGHYVMVSPGYAQTNEKWFPRANEFDPHRWDEETSSNIDTDAVDYGFGKVTKGVSSPYLPFGGGRHRCIGEQFAYVQLGTILATYVYNIKWRFKKDGSLPPVDYQSMVTLPMEPAEIEWEKRETCVY |
| V130W | -10.6 | -- | MALKDCIVDVVDRFSALPVPVKLAVLILVPIVYNLVWQFVYSLRKDRAPLVFHWVPWVGSAVVYGMQPYQFFESCREKYGDVFAFVMLGKVMTVYLGPKGHEFVLNAKLADVSAEAAYSHLTTPVFGKGWIFDCPNSRLMEQKKFAKTALTKEAFQRYVPRIQEEVLDYFKACSQFRMNERNNGVANVMKTQPEMTILTASKSLMGDDMRARFDASFAKLYSDLDKGFTPINFVFPHLPLPAYWKRDAAQQKISATYMSLINERRKTGDIVPDRDLIDSLMTNSTYKDGVKMTDQEVANLLIGVLMGGQHTSASTSAWFLLHLAEQPKLQEELYSEVLSVLADKGGSLKDLAYDDLQKMPLINQTIKETLRLHMPLHSIFRKVMNPLVVPNTKYVVPKGHYVMVSPGYAQTNEKWFPRANEFDPHRWDEETSSNIDTDAVDYGFGKVTKGVSSPYLPFGGGRHRCIGEQFAYVQLGTILATYVYNIKWRFKKDGSLPPVDYQSMVTLPMEPAEIEWEKRETCVY |
| I131W | -10.5 | -- | MALKDCIVDVVDRFSALPVPVKLAVLILVPIVYNLVWQFVYSLRKDRAPLVFHWVPWVGSAVVYGMQPYQFFESCREKYGDVFAFVMLGKVMTVYLGPKGHEFVLNAKLADVSAEAAYSHLTTPVFGKGVWFDCPNSRLMEQKKFAKTALTKEAFQRYVPRIQEEVLDYFKACSQFRMNERNNGVANVMKTQPEMTILTASKSLMGDDMRARFDASFAKLYSDLDKGFTPINFVFPHLPLPAYWKRDAAQQKISATYMSLINERRKTGDIVPDRDLIDSLMTNSTYKDGVKMTDQEVANLLIGVLMGGQHTSASTSAWFLLHLAEQPKLQEELYSEVLSVLADKGGSLKDLAYDDLQKMPLINQTIKETLRLHMPLHSIFRKVMNPLVVPNTKYVVPKGHYVMVSPGYAQTNEKWFPRANEFDPHRWDEETSSNIDTDAVDYGFGKVTKGVSSPYLPFGGGRHRCIGEQFAYVQLGTILATYVYNIKWRFKKDGSLPPVDYQSMVTLPMEPAEIEWEKRETCVY |
| P135W | -10.7 | Hydrogen→ 4
|
MALKDCIVDVVDRFSALPVPVKLAVLILVPIVYNLVWQFVYSLRKDRAPLVFHWVPWVGSAVVYGMQPYQFFESCREKYGDVFAFVMLGKVMTVYLGPKGHEFVLNAKLADVSAEAAYSHLTTPVFGKGVIFDCWNSRLMEQKKFAKTALTKEAFQRYVPRIQEEVLDYFKACSQFRMNERNNGVANVMKTQPEMTILTASKSLMGDDMRARFDASFAKLYSDLDKGFTPINFVFPHLPLPAYWKRDAAQQKISATYMSLINERRKTGDIVPDRDLIDSLMTNSTYKDGVKMTDQEVANLLIGVLMGGQHTSASTSAWFLLHLAEQPKLQEELYSEVLSVLADKGGSLKDLAYDDLQKMPLINQTIKETLRLHMPLHSIFRKVMNPLVVPNTKYVVPKGHYVMVSPGYAQTNEKWFPRANEFDPHRWDEETSSNIDTDAVDYGFGKVTKGVSSPYLPFGGGRHRCIGEQFAYVQLGTILATYVYNIKWRFKKDGSLPPVDYQSMVTLPMEPAEIEWEKRETCVY |
| N136C | -10.2 | -- | MALKDCIVDVVDRFSALPVPVKLAVLILVPIVYNLVWQFVYSLRKDRAPLVFHWVPWVGSAVVYGMQPYQFFESCREKYGDVFAFVMLGKVMTVYLGPKGHEFVLNAKLADVSAEAAYSHLTTPVFGKGVIFDCPCSRLMEQKKFAKTALTKEAFQRYVPRIQEEVLDYFKACSQFRMNERNNGVANVMKTQPEMTILTASKSLMGDDMRARFDASFAKLYSDLDKGFTPINFVFPHLPLPAYWKRDAAQQKISATYMSLINERRKTGDIVPDRDLIDSLMTNSTYKDGVKMTDQEVANLLIGVLMGGQHTSASTSAWFLLHLAEQPKLQEELYSEVLSVLADKGGSLKDLAYDDLQKMPLINQTIKETLRLHMPLHSIFRKVMNPLVVPNTKYVVPKGHYVMVSPGYAQTNEKWFPRANEFDPHRWDEETSSNIDTDAVDYGFGKVTKGVSSPYLPFGGGRHRCIGEQFAYVQLGTILATYVYNIKWRFKKDGSLPPVDYQSMVTLPMEPAEIEWEKRETCVY |
| Y118W | -10.7 | -- | MALKDCIVDVVDRFSALPVPVKLAVLILVPIVYNLVWQFVYSLRKDRAPLVFHWVPWVGSAVVYGMQPYQFFESCREKYGDVFAFVMLGKVMTVYLGPKGHEFVLNAKLADVSAEAAWSHLTTPVFGKGVIFDCPNSRLMEQKKFAKTALTKEAFQRYVPRIQEEVLDYFKACSQFRMNERNNGVANVMKTQPEMTILTASKSLMGDDMRARFDASFAKLYSDLDKGFTPINFVFPHLPLPAYWKRDAAQQKISATYMSLINERRKTGDIVPDRDLIDSLMTNSTYKDGVKMTDQEVANLLIGVLMGGQHTSASTSAWFLLHLAEQPKLQEELYSEVLSVLADKGGSLKDLAYDDLQKMPLINQTIKETLRLHMPLHSIFRKVMNPLVVPNTKYVVPKGHYVMVSPGYAQTNEKWFPRANEFDPHRWDEETSSNIDTDAVDYGFGKVTKGVSSPYLPFGGGRHRCIGEQFAYVQLGTILATYVYNIKWRFKKDGSLPPVDYQSMVTLPMEPAEIEWEKRETCVY |
| C134W | -10.7 | -- | MALKDCIVDVVDRFSALPVPVKLAVLILVPIVYNLVWQFVYSLRKDRAPLVFHWVPWVGSAVVYGMQPYQFFESCREKYGDVFAFVMLGKVMTVYLGPKGHEFVLNAKLADVSAEAAYSHLTTPVFGKGVIFDWPNSRLMEQKKFAKTALTKEAFQRYVPRIQEEVLDYFKACSQFRMNERNNGVANVMKTQPEMTILTASKSLMGDDMRARFDASFAKLYSDLDKGFTPINFVFPHLPLPAYWKRDAAQQKISATYMSLINERRKTGDIVPDRDLIDSLMTNSTYKDGVKMTDQEVANLLIGVLMGGQHTSASTSAWFLLHLAEQPKLQEELYSEVLSVLADKGGSLKDLAYDDLQKMPLINQTIKETLRLHMPLHSIFRKVMNPLVVPNTKYVVPKGHYVMVSPGYAQTNEKWFPRANEFDPHRWDEETSSNIDTDAVDYGFGKVTKGVSSPYLPFGGGRHRCIGEQFAYVQLGTILATYVYNIKWRFKKDGSLPPVDYQSMVTLPMEPAEIEWEKRETCVY |
| R381W | -9.8 | -- | MALKDCIVDVVDRFSALPVPVKLAVLILVPIVYNLVWQFVYSLRKDRAPLVFHWVPWVGSAVVYGMQPYQFFESCREKYGDVFAFVMLGKVMTVYLGPKGHEFVLNAKLADVSAEAAYSHLTTPVFGKGVIFDCPNSRLMEQKKFAKTALTKEAFQRYVPRIQEEVLDYFKACSQFRMNERNNGVANVMKTQPEMTILTASKSLMGDDMRARFDASFAKLYSDLDKGFTPINFVFPHLPLPAYWKRDAAQQKISATYMSLINERRKTGDIVPDRDLIDSLMTNSTYKDGVKMTDQEVANLLIGVLMGGQHTSASTSAWFLLHLAEQPKLQEELYSEVLSVLADKGGSLKDLAYDDLQKMPLINQTIKETLRLHMPLHSIFWKVMNPLVVPNTKYVVPKGHYVMVSPGYAQTNEKWFPRANEFDPHRWDEETSSNIDTDAVDYGFGKVTKGVSSPYLPFGGGRHRCIGEQFAYVQLGTILATYVYNIKWRFKKDGSLPPVDYQSMVTLPMEPAEIEWEKRETCVY |
| R381S | -10.3 | -- | MALKDCIVDVVDRFSALPVPVKLAVLILVPIVYNLVWQFVYSLRKDRAPLVFHWVPWVGSAVVYGMQPYQFFESCREKYGDVFAFVMLGKVMTVYLGPKGHEFVLNAKLADVSAEAAYSHLTTPVFGKGVIFDCPNSRLMEQKKFAKTALTKEAFQRYVPRIQEEVLDYFKACSQFRMNERNNGVANVMKTQPEMTILTASKSLMGDDMRARFDASFAKLYSDLDKGFTPINFVFPHLPLPAYWKRDAAQQKISATYMSLINERRKTGDIVPDRDLIDSLMTNSTYKDGVKMTDQEVANLLIGVLMGGQHTSASTSAWFLLHLAEQPKLQEELYSEVLSVLADKGGSLKDLAYDDLQKMPLINQTIKETLRLHMPLHSIFSKVMNPLVVPNTKYVVPKGHYVMVSPGYAQTNEKWFPRANEFDPHRWDEETSSNIDTDAVDYGFGKVTKGVSSPYLPFGGGRHRCIGEQFAYVQLGTILATYVYNIKWRFKKDGSLPPVDYQSMVTLPMEPAEIEWEKRETCVY |
| R381T | -10.0 | -- | MALKDCIVDVVDRFSALPVPVKLAVLILVPIVYNLVWQFVYSLRKDRAPLVFHWVPWVGSAVVYGMQPYQFFESCREKYGDVFAFVMLGKVMTVYLGPKGHEFVLNAKLADVSAEAAYSHLTTPVFGKGVIFDCPNSRLMEQKKFAKTALTKEAFQRYVPRIQEEVLDYFKACSQFRMNERNNGVANVMKTQPEMTILTASKSLMGDDMRARFDASFAKLYSDLDKGFTPINFVFPHLPLPAYWKRDAAQQKISATYMSLINERRKTGDIVPDRDLIDSLMTNSTYKDGVKMTDQEVANLLIGVLMGGQHTSASTSAWFLLHLAEQPKLQEELYSEVLSVLADKGGSLKDLAYDDLQKMPLINQTIKETLRLHMPLHSIFTKVMNPLVVPNTKYVVPKGHYVMVSPGYAQTNEKWFPRANEFDPHRWDEETSSNIDTDAVDYGFGKVTKGVSSPYLPFGGGRHRCIGEQFAYVQLGTILATYVYNIKWRFKKDGSLPPVDYQSMVTLPMEPAEIEWEKRETCVY |
| D133W | -10.0 | -- | MALKDCIVDVVDRFSALPVPVKLAVLILVPIVYNLVWQFVYSLRKDRAPLVFHWVPWVGSAVVYGMQPYQFFESCREKYGDVFAFVMLGKVMTVYLGPKGHEFVLNAKLADVSAEAAYSHLTTPVFGKGVIFWCPNSRLMEQKKFAKTALTKEAFQRYVPRIQEEVLDYFKACSQFRMNERNNGVANVMKTQPEMTILTASKSLMGDDMRARFDASFAKLYSDLDKGFTPINFVFPHLPLPAYWKRDAAQQKISATYMSLINERRKTGDIVPDRDLIDSLMTNSTYKDGVKMTDQEVANLLIGVLMGGQHTSASTSAWFLLHLAEQPKLQEELYSEVLSVLADKGGSLKDLAYDDLQKMPLINQTIKETLRLHMPLHSIFRKVMNPLVVPNTKYVVPKGHYVMVSPGYAQTNEKWFPRANEFDPHRWDEETSSNIDTDAVDYGFGKVTKGVSSPYLPFGGGRHRCIGEQFAYVQLGTILATYVYNIKWRFKKDGSLPPVDYQSMVTLPMEPAEIEWEKRETCVY |
| P135H | -10.7 | -- | MALKDCIVDVVDRFSALPVPVKLAVLILVPIVYNLVWQFVYSLRKDRAPLVFHWVPWVGSAVVYGMQPYQFFESCREKYGDVFAFVMLGKVMTVYLGPKGHEFVLNAKLADVSAEAAYSHLTTPVFGKGVIFDCHNSRLMEQKKFAKTALTKEAFQRYVPRIQEEVLDYFKACSQFRMNERNNGVANVMKTQPEMTILTASKSLMGDDMRARFDASFAKLYSDLDKGFTPINFVFPHLPLPAYWKRDAAQQKISATYMSLINERRKTGDIVPDRDLIDSLMTNSTYKDGVKMTDQEVANLLIGVLMGGQHTSASTSAWFLLHLAEQPKLQEELYSEVLSVLADKGGSLKDLAYDDLQKMPLINQTIKETLRLHMPLHSIFRKVMNPLVVPNTKYVVPKGHYVMVSPGYAQTNEKWFPRANEFDPHRWDEETSSNIDTDAVDYGFGKVTKGVSSPYLPFGGGRHRCIGEQFAYVQLGTILATYVYNIKWRFKKDGSLPPVDYQSMVTLPMEPAEIEWEKRETCVY |
| P135A | -10.8 | Hydrophobic→ 5
|
MALKDCIVDVVDRFSALPVPVKLAVLILVPIVYNLVWQFVYSLRKDRAPLVFHWVPWVGSAVVYGMQPYQFFESCREKYGDVFAFVMLGKVMTVYLGPKGHEFVLNAKLADVSAEAAYSHLTTPVFGKGVIFDCANSRLMEQKKFAKTALTKEAFQRYVPRIQEEVLDYFKACSQFRMNERNNGVANVMKTQPEMTILTASKSLMGDDMRARFDASFAKLYSDLDKGFTPINFVFPHLPLPAYWKRDAAQQKISATYMSLINERRKTGDIVPDRDLIDSLMTNSTYKDGVKMTDQEVANLLIGVLMGGQHTSASTSAWFLLHLAEQPKLQEELYSEVLSVLADKGGSLKDLAYDDLQKMPLINQTIKETLRLHMPLHSIFRKVMNPLVVPNTKYVVPKGHYVMVSPGYAQTNEKWFPRANEFDPHRWDEETSSNIDTDAVDYGFGKVTKGVSSPYLPFGGGRHRCIGEQFAYVQLGTILATYVYNIKWRFKKDGSLPPVDYQSMVTLPMEPAEIEWEKRETCVY |
Docked WT:
Docked Y132F Variant:
Docked P135W Secondary Rescue Mutation:
Ramachadaran Plot:
 Ramachandran plot analysis of the P135A variant indicates preserved backbone stability and proper structural geometry. The majority of residues fall within favoured and allowed regions, forming dense clusters in the characteristic α-helix (φ ≈ −60°, ψ ≈ −45°) and β-sheet (φ ≈ −120°, ψ ≈ 120°) conformational zones. Very few outliers are present, suggesting the absence of significant backbone strain or modelling artifacts. Inherently flexible glycine residues display broader dispersion but remain within acceptable contour regions, indicating normal local flexibility rather than instability. Valine and isoleucine residues show tight clustering within favoured zones, supporting maintenance of core packing and steric integrity. Pre-proline, trans-proline, and cis-proline residues appear in expected conformational regions without abnormal torsional deviations. Overall, the plot demonstrates that the P135A substitution did not introduce global unfolding, compensatory distortions, or abnormal peptide bond geometry, supporting the structural stability of the modified protein.
Ramachandran plot analysis of the P135A variant indicates preserved backbone stability and proper structural geometry. The majority of residues fall within favoured and allowed regions, forming dense clusters in the characteristic α-helix (φ ≈ −60°, ψ ≈ −45°) and β-sheet (φ ≈ −120°, ψ ≈ 120°) conformational zones. Very few outliers are present, suggesting the absence of significant backbone strain or modelling artifacts. Inherently flexible glycine residues display broader dispersion but remain within acceptable contour regions, indicating normal local flexibility rather than instability. Valine and isoleucine residues show tight clustering within favoured zones, supporting maintenance of core packing and steric integrity. Pre-proline, trans-proline, and cis-proline residues appear in expected conformational regions without abnormal torsional deviations. Overall, the plot demonstrates that the P135A substitution did not introduce global unfolding, compensatory distortions, or abnormal peptide bond geometry, supporting the structural stability of the modified protein.
 Whole-protein RMSD values remain below 0.1 Å across all comparisons, confirming that global protein architecture is preserved. The Y132F resistance mutation shows minimal ligand displacement (0.2 Å) despite reduced binding affinity, indicating that subtle local geometric distortions — not large-scale structural shifts — underlie resistance. The P135A rescue mutation maintains similar structural stability (0.3 Å ligand shift) while restoring affinity, supporting a geometry-driven recovery mechanism. In contrast, the bulky P135W substitution causes dramatic ligand reorientation (7.0 Å shift), demonstrating that steric disruption of binding pocket geometry severely compromises docking stability. Collectively, these findings establish that precise spatial alignment — rather than overall structural change or increased contact count — governs fluconazole binding efficacy. All structures maintain near-identical global architecture (RMSD < 0.1 Å), confirming resistance is not due to protein unfolding. Minimal ligand shifts in Y132F (0.2 Å) despite reduced affinity indicate that subtle active-site geometric distortions drive binding loss. In contrast, the bulky P135W mutation causes major ligand displacement (7.0 Å), demonstrating that binding stability is governed by precise spatial alignment rather than contact quantity. In essence, binding affinity correlates with geometric alignment, not global structure or interaction count. Subtle local distortions weaken binding, while steric disruption causes complete ligand displacement. Bulky, contact-focused mutation, P135W, increased interactions but compromised binding geometry through steric interference. It flipped fluconazole 180 degrees to compensate for backbone strain
Whole-protein RMSD values remain below 0.1 Å across all comparisons, confirming that global protein architecture is preserved. The Y132F resistance mutation shows minimal ligand displacement (0.2 Å) despite reduced binding affinity, indicating that subtle local geometric distortions — not large-scale structural shifts — underlie resistance. The P135A rescue mutation maintains similar structural stability (0.3 Å ligand shift) while restoring affinity, supporting a geometry-driven recovery mechanism. In contrast, the bulky P135W substitution causes dramatic ligand reorientation (7.0 Å shift), demonstrating that steric disruption of binding pocket geometry severely compromises docking stability. Collectively, these findings establish that precise spatial alignment — rather than overall structural change or increased contact count — governs fluconazole binding efficacy. All structures maintain near-identical global architecture (RMSD < 0.1 Å), confirming resistance is not due to protein unfolding. Minimal ligand shifts in Y132F (0.2 Å) despite reduced affinity indicate that subtle active-site geometric distortions drive binding loss. In contrast, the bulky P135W mutation causes major ligand displacement (7.0 Å), demonstrating that binding stability is governed by precise spatial alignment rather than contact quantity. In essence, binding affinity correlates with geometric alignment, not global structure or interaction count. Subtle local distortions weaken binding, while steric disruption causes complete ligand displacement. Bulky, contact-focused mutation, P135W, increased interactions but compromised binding geometry through steric interference. It flipped fluconazole 180 degrees to compensate for backbone strain
 Increased intermolecular contacts in the Y132F and P135W mutants did not translate to improved binding affinity, demonstrating that interaction quantity alone does not determine docking stability. The P135A substitution restored binding through improved geometric alignment rather than increased contact density.
Increased intermolecular contacts in the Y132F and P135W mutants did not translate to improved binding affinity, demonstrating that interaction quantity alone does not determine docking stability. The P135A substitution restored binding through improved geometric alignment rather than increased contact density.
Conclusion
Conclusion
This project began with a straightforward objective: improve docking affinity by increasing molecular interactions. Early secondary mutations near the Y132F resistance site successfully increased contact counts and, in some cases, produced binding scores approaching wild-type levels. However, a consistent plateau emerged. Variants with stronger interaction numbers still failed to restore proper ligand positioning. The turning point occurred with the P135W mutation. Although it introduced additional contacts, it forced fluconazole to rotate nearly 180° within the binding pocket to accommodate steric bulk. Energetically, the docking score appeared competitive. Geometrically, however, the ligand adopted a strained conformation. This revealed a critical insight: interaction quantity does not equal binding quality.
The resistance phenotype appeared not to be driven primarily by loss of contacts, but by distortion of three-dimensional binding architecture. Larger, bulkier substitutions generated favourable interactions yet imposed rigidity and spatial strain, ultimately weakening true binding complementarity. The Y132F mutation likely altered pocket symmetry in a way that required adaptive flexibility rather than additional steric forcing. This realization shifted the strategy entirely. Proline at position 135 was identified as a rigid structural constraint adjacent to the resistance site. Known for introducing backbone kinks and limiting conformational freedom, proline may have prevented the binding pocket from accommodating the geometric disturbance caused by Y132F. Replacing rigidity with additional bulky residues only compounded the problem. The solution was not to add structure-- it was to remove constraint. Alanine was selected because of its minimal, nonreactive side chain and its established role in alanine-scanning mutagenesis, where it is used specifically to remove side-chain influence while preserving backbone integrity. Substituting P135 with alanine reduced steric rigidity without introducing artificial interactions. Unlike previous mutations that forced fluconazole into position, P135A allowed the binding pocket to relax and re-symmetrize. The ligand adopted a lower-strain orientation, and binding affinity was partially restored without introducing backbone instability, as confirmed by Ramachandran analysis.
The central discovery of this project is that antifungal resistance may be fundamentally geometric rather than purely chemical. Resistance does not always arise from the loss of interaction strength; it can emerge from subtle architectural distortion and reduced conformational adaptability. By restoring flexibility rather than maximizing contacts, binding geometry can be improved even in the presence of a resistance mutation.
Implication and Future Directions
This has broader implications. If small structural adjustments can partially restore drug binding, then resistance may not always require entirely new antifungal drugs. Instead of replacing fluconazole, we may be able to redesign it, or design derivatives that exploit compensatory structural features within resistant enzymes. In low-resource settings where access to new antifungals is limited, extending the functional lifespan of existing drugs could have a substantial clinical impact. Future work should experimentally validate the predicted compensatory mutation by introducing P135A into fluconazole-resistant Candida auris strains and measuring changes in minimum inhibitory concentration. Structural studies such as molecular dynamics simulations could further evaluate binding pocket flexibility and conformational dynamics. Ultimately, these findings support a structure-guided framework for resistance modulation, where understanding geometric disruption informs targeted drug redesign rather than drug replacement. This project reframes antifungal resistance not as an irreversible endpoint, but as a structural imbalance that may be corrected. By shifting the focus from interaction maximization to geometric restoration, it demonstrates that even a single amino acid-- chosen strategically-- can reshape how a life-saving drug binds.
Citations
Below are the primary and most essential sources used in this research.
For the complete list of references, please refer to my project logbook.
---
Blood. (2015). Secondary mutations as mediators of resistance to targeted therapies. Blood, 125(21), 3236–3244. https://ashpublications.org/blood/article/125/21/3236/34001/Secondary-mutations-as-mediators-of-resistance-to
Proceedings of the National Academy of Sciences of the United States of America. (2001). A secondary drug resistance mutation of TEM-1 β-lactamase that suppresses misfolding and aggregation. PNAS, 98(1), 283–288. https://www.pnas.org/doi/abs/10.1073/pnas.98.1.283
Sanger Institute. (n.d.). ‘Rescue mutations’ that suppress harmful DNA changes could shed light on the origins of genetic disorders. https://www.sanger.ac.uk/news_item/rescue-mutations-that-suppress-harmful-dna-changes-could-shed-light-on-origins-of-genetic-disorders/
ScienceDirect. (2019). Candida auris and antifungal resistance mechanisms. Current Opinion in Microbiology. https://www.sciencedirect.com/science/article/abs/pii/S1087184519301501
ScienceDirect. (2022). Mechanisms of antifungal resistance in Candida auris. Current Opinion in Microbiology. https://www.sciencedirect.com/science/article/pii/S1369527422001217
UniProt Consortium. (n.d.). UniProt: The universal protein knowledgebase. https://www.uniprot.org/
YouTube. (n.d.). [Video on protein structure/docking – IvLI9ECRMPI]. YouTube. https://www.youtube.com/watch?v=IvLI9ECRMPI
YouTube. (n.d.). [Video on protein modelling – eLy7PdzRgLs]. YouTube. https://www.youtube.com/watch?v=eLy7PdzRgLs
Chen, J., Sawyer, N., & Regan, L. (1992). Rapid mapping of protein functional epitopes by combinatorial alanine scanning. Proceedings of the National Academy of Sciences of the United States of America, 89(20), 9735–9739. https://www.pnas.org/doi/10.1073/pnas.160252097
LibreTexts. (n.d.). Van der Waals interactions. Chemistry LibreTexts. https://chem.libretexts.org/Bookshelves/Physical_and_Theoretical_Chemistry_Textbook_Maps/Supplemental_Modules_(Physical_and_Theoretical_Chemistry)/Physical_Properties_of_Matter/Atomic_and_Molecular_Properties/Intermolecular_Forces/Specific_Interactions/Van_Der_Waals_Interactions
Meme Suite. (n.d.). Alphabet's documentation. https://meme-suite.org/meme/doc/alphabets.html
National Center for Biotechnology Information. (2021). Cation–π interactions in protein structure and function. Journal of Molecular Biology. https://pmc.ncbi.nlm.nih.gov/articles/PMC8338773/
WHO. (n.d.). Global antifungal resistance and Candida auris report. World Health Organization. https://iris.who.int/server/api/core/bitstreams/b2fa3f20-191e-4112-ab2d-fb0f56fa8fd2/content
YouTube. (n.d.). [Protein modelling tutorial – i3cdgCpCmvU]. YouTube. https://www.youtube.com/watch?v=i3cdgCpCmvU
YouTube. (n.d.). [Molecular docking tutorial – 5_WQaLXcc18]. YouTube. https://www.youtube.com/watch?v=5_WQaLXcc18
CB-Dock2. (n.d.). CB-Dock2: Protein–ligand blind docking server. https://cadd.labshare.cn/cb-dock2/php/blinddock.php
Acknowledgement
I sincerely thank my science fair teacher, Ms. O, for answering my relentless questions. You made my first science fair competition experience so memorable.
I’d like to extend my gratitude to my Biology Honours teacher, Mr. H, for allowing me to come to you regarding every obstacle and issue I faced along this tumultuous research journey (by the way, I still don’t know how to code…). Your humour towards my frustration always made me feel better, and your unwavering optimism towards me and my abilities is the reason why I have this project here today.
And of course, to my family: thank you for always supporting me, listening to every rant and brainstorm about this project, and encouraging me when the science felt overwhelming. Your patience, love, and belief in me made this journey possible, and I am grateful for every pep talk and word of encouragement.