Innovation for Treatments on Creutzfeldt-Jakob Disease
Zimmel Nadir
FFCA High School Campus
Grade 11
Presentation
No video provided
Problem
Creutzfeldt-Jakob disease is a prion disease, characterized by the misfolding of proteins in the brain, which leads to rapid neurodegeneration. These proteins, turned prions, are unable to be recognized by the immune system as a threat, can resist enzymes that break them down, and have no effective medication or treatment as of today. Not only that, but the detection of the disease is also extremely difficult, as symptoms can appear decades after a patient has contracted the disease. In most cases, the cause of protein aggregation in a patients brain is also unknown. The intricacies of CJD make diagnosis and treatment remarkably difficult, meaning that the majority of patients diagnosed with CJD die within the first year.
The goal of this project is to innovate a method in which CJD can be treated after protein aggregation in the brain has been detected, but before conditions in a patient become critical. This task leads to a few crucial questions.
- Once misfolding is detected, is it possible to undo that aggregation?
- How can we study more accessible organisms, such as yeast, to make new discoveries on prions?
- Can that knowledge be used to progress treatment for CJD?
Method
Brief Summary of Background Knowledge
Most studies focus on handling the prion protein or PrP^c before it misfolds into the actual PrP^Sc form. This is done through things like gene regulation/expression, epigenetics, and genetic editing (CHARM, CRISPR, etc). This is a plausible and understandable solution, as when the prion protein actually misfolds, there isn't a lot you can necessarily do to treat the disease since unfolding it within the brain is risky and difficult. However CJD (the most common prion disease in humans) is often is sporadic (85%), meaning it is contracted spontaneously. So there often is no way to know if a person will contract CJD before symptoms, which can get confused with other disease like alzheimer's or dementia. Because of this, conditions in a patient become critical, and they are most likely to pass away that same year.
The Goal
What I am trying to accomplish with my experiment is to deal with PrP^Sc once a patient has already contracted it. Meaning the proteins have already misfolded, and the patient has been diagnosed. Is there a way to unfold this prion, even with the fragility of the brain? Can we stop it from spreading/propagating entirely? ___________________________________________________________________________________________________________________________
Using Yeast
- Prions are not only found in animals (where they can cause extremely harmful neurodegenerative diseases). They can also be found in fungi such as yeast (Saccharomyces cerevisiae)
- Like human prions, these fungal prions are also able to change their conformation on their own terms. Adopting multiple shapes/conformations, and causing other proteins to do the same.
- Animals such as humans do not have the certain chaperone Hsp 104, but we do have others such as Hsp 40, 70 which all help with the disaggregation of prions by stopping them from misfolding in the first place (but it is not foolproof, which can lead to CJD)
- Hsp104 is able to be introduced to cells it usually doesn't belong to, such as mammalian cells (making it exogenous Hsp 104)
- Exogeneous Hsp104 is able to team up with these other mammalian chaperones (Hsp 40, 70) and promote the disaggregation of proteins in humans.
- This process has veen thought to help with treating diseases associated with amyloid diseases (defined as diseases which misfolded proteins build up as plaque in an organ)
- But there is still doubt that this would work at all, as scientists worry that Hsp104 alone may not be enough. These limitations may mean that other treatments will have to be used alongside this introduction of a fungal chaperone.
- The study was to show and explain findings on how certain chaperones in fungi and humans can work together, and then perhaps lead to certain therapeutic treatments for certain diseases associated with them (amyloid diseases, such as CJD)
Hsp104 is a chaperone (which is defined as a protein which helps the other proteins) specifically to fungi such as yeast. This chaperone is interesting, because rather than preventing the misfolding of proteins like chaperones Hsp40 and 70, by working together, sharing information, etc, it takes the already aggregated proteins and helps it to unfold and keep the fungi safe. ___________________________________________________________________________________________________________________________
Hsp104 - A Molecular Chaperone Protein (In Yeast)
- A high shock protein (meaning they react to external factors/stressors such as heat, ethanol, and sodium arsenite)
- Described as a disaggregase (a molecular chaperone behaving like an enzyme to break down/reverse the aggregation of proteins, making them functional)
- It works with/ cooperates with Ydj1p (Hsp40) and Ssa1p (Hsp70) to refold/reacitivate protein aggregates
- The stressors of heat, ethanol, and sodium arsenite (and of course others) make it so proteins become tangled. Hsp 104 helps to detangle them, turning them into a liquid form so they can disperse and function properly
Hsp 104 has been studied in yeast models for human diseases, such as Creutzfeldt-Jakob disease (and Huntingson disease) In YEAST there are three types of proteins strains, and three related proteins
[PSI+] : Related to the protein Sup35p (impacts how genetic information is expressed) [PIN+]: Related to the protein Rnq1p (similar to PSI) [URE3] : Related to the protein Ure2 (regulates nitrogen metabolism)
- If HSP104 is deleted, these prion strains are not able to be multiplied (propagated) .
- In order for that prion to be inherited by a daughter cell, it must be broken down. Without that fragmentation, the aggregate becomes large and is not able to be passed to the daughter cell effectively
- With Hsp40 and Hsp70 does Hsp104 break down these strains into smaller fragments. Those smaller fragments go on to convert other proteins into prions
- If HSP is underexpressed/deleted, that break down can’t happen, preventing the transfer of those prions (however, its a double edged sword, because a large toxic aggregate can still have negative impacts on those yeast cells)
- If HSP104 is overexpressed, it can cure the yeast cells of the [PSI+] prion strain (reduces clumping/protein aggregation because it just breaks it down so well)
- In mice that were genetically modified to give Huntington's disease, extra HSP104 let them live longer. (what could that mean for humans?)
In YEAST, you want a balance of Hsp104, as you don’t want the prion strain to go extinct in the yeast cell because while they are harmful, they help yeast cells adapt and thrive in very harsh environmental stress. So a “goldilocks” amount of HSP104 ensures it stays regulated, but not extinct.
Btn2/Cur1 can help heal the prion strain URE3. These two proteins recognise misfolded prions, and target them to prevent their accumulation, and therefore their spread. They do this through DEGRADING the protein, or INHIBITING THE PROPAGATION. ___________________________________________________________________________________________________________________________
Final Method
If we take these Btn2/Cur1 proteins, and insert them exogenously with a system that has been ridden with misfolded proteins, would the introduction of these proteins fight against that propagation and degrade the protein, therefore ceasing the aggregation? And if those proteins are stop forming aggregates, would the disease be silenced?
The concept of introducing something from outside its system has been thought of with the chaperone HSP104, by having mammalian chaperones work together and fight prion misfolding. Could it not be done with other structural elements of the fungi? Using what we know about the HSP104 chaperone, could the protein Btn2/Cur1 be used the same way?
QUESTION- Can the protein Btn2 and Cur1 be used exogenously like the chaperone protein Hsp104 to cease the aggregation of the prion protein, therefore stopping or slowing neurodegeneration?
Research
BACKGROUND RESEARCH
What is Creutzfeldt-Jakob Disease?
- A rare brain disorder (any condition which impairs the brain's abilities) which leads to dementia (a general term to describe memory loss, which affects the daily lives of an individual)
- Belongs to a group known as PRION DISORDERS*
- Similar to alzeimers, but it moves faster and is fatal (typically within a year a person diagnosed with CJD will pass away) This is often due to the issues which come from the disease, such as trouble eating, heart and lung failure, pneumonia, and other infections which can develop
- SYMPTOMS INCLUDE; changes in personality, loss of memory, blurry vision, insomnia, trouble talking and coordinating oneself. Dizziness, slurred speech, hallucinations, depression, withdrawal, anxiety, irritability.
- In the final stage the patient will be bedridden, the complete loss of speech, and an inevitable death.
- There is nothing which can be done at this stage in the disease (however advancements in end of life care, which focus on incurable diseases, mean the patients go out peacefully)
- A change in mental ability may be noticeable in the early stages, and later on dementia can develop
- As we know CJD and prion diseases alike so far have no known cure for these types of diseases. (prion diseases are also called transmissible spongiform encephalopathies)
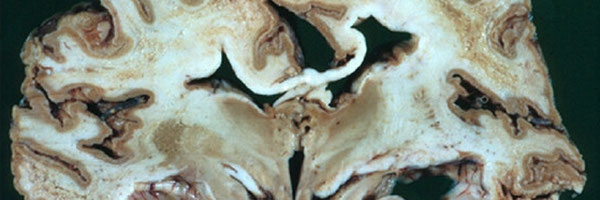
- SPONGIFORM refers to how the brain looks like a sponge when affected with a prion disease, because of all the holes
- Like prion diseases, CJD creates issues around memory and coordination, often leading to dementia and, inevitably, death
- 70% of individuals will die within the first year of contracting the disease
Types of CJD
SPORADIC CJD
- Most common
- Cause is unclear and seemingly spontaneous , but is suspected to be linked to, again, a brain protein folding which turns into a PRION*
- Ages range from 45-75
VARIANT CJD
- Suspected to be caused by consuming meat from a cow which has bovine spongiform encephalopathy (BSE, or mad cow disease) It is a similar prion disease
- To prevent it, there have been strict controls on meat and its protection from diseases such as BSE
- Can also occur through blood transfusion
- The time it takes for the disease to develop after initial contact with the infected meat, is again unclear FAMILIAL / INHERITED
- A rare genetic condition where an individual will inherit a mutated prion protein from their parent, causing prion formation in adulthood, and the potential for CJD
- The symptoms typically develop in adults in their early 50s.
- Genetic mutation IATROGENIC CJD
- The infection is spread accidentally from someone with CJD, through medical/surgical treatments
- Iatrogenic CJD had been spread in the past through pituitary growth hormones extracted from people with this disease (it is now synthetically made, so no there is no risk)
- Can also occur if surgical instruments aren't properly cleaned after being used on an individual with CJD, and then had been used on someone else
- Increased awareness has reduced this type of CJD however
IMPORTANT VOCABULARY prion/ prion disease- A certain misshapen protein in the brain which seems to be the cause of CJD // a prion is a misshapen and misfolded protein which causes other proteins to misfold, and thus creating clumps of prions which results in brain damage Sporadic- the most common type of CJD (85%) , and develops suddenly Variant- A type of CJD which results in digesting cattle infected with mad cow disease\, another smilat prion disease Familial- a rare genetic mutation which results in the transfer of mutated proteins from a parent, which result then in symptoms of CJD Iatrogenic- a type of CJD in which the disease is spread accidentally from someone with it, through a medical procedure genetic CJD- a type of CJD where the genes mutate, thus resulting in messed up proteins, which then in turn result in prions.
How does CJD occur? // What is a Prion Disease, and how does it occur?
- CJD is caused by infections proteins called PRIONS in the brain
- Proteins are molecules of amino acids which help cells function. They begin as a string of said amino acids, and fold over in order to perform their functions.
The Genetics (specifically for genetic CJD)
- Chromosome 20 (as we know we have 46 chromosomes in total, and 23 pairs), where the prior protein is, and all genetic prion disease result from mutations in this gene
- The gene on chromosome 20 is called PRNP, and encodes for a prion protein
- RNA (translated from DNA) will communicate to the genes inside a cell's nucleus on what proteins it should make (proteins make the structure and functions of the human body)
- The nucleotides on RNA and DNA (the rungs on the ladder) make up the alphabet of our genetic code.
- Read in three letter pairs, which in turn make an amino acid, which in turn make a protein
- As we know, chromosomes contain DNA which contain genetic information. In order to build who you are, these three things work together.
- The GENES hold specific parts of your DNA. That DNA will send messages by RNA (mRNA), to create PROTEINS.
- If there is a genetic mutation then it can result in wonky and folded proteins or harmful PRIONS.
How do proteins and prions fold?
- We start with a normal protein, which attains a molten globule state (like partially folded), and continues to fold until it reaches its normal end stage.
- But during the folding process errors can occur (that genetic mutation) and those proteins become misshapen
- But not to worry, these proteins are sent for repair, and gets corrected so it can become normal
- But if it does not, it goes to proteases to wear down, and get recycled into the body.
- BUT, if the misfolded protein can since changed into a PRION, it resists the proteases and can not be broken down.
- This is due to the structure of said prion. A regular prion protein will have 43% alpha helices and 03% beta sheets. But a prion will have 40% alpha helices and 43% beta sheets. This difference results in that resistant (protease resistant)
- These undegraded proteins are then called PRIONS
- And then these prions can go and affect other prion proteins to misfold as well.
The Deal With Prions
- There are normal and harmless prions, which are technically just folded proteins, and they are found in all parts of the body, especially the brain. Their cause is still unclear, but it is speculated to be responsible for transporting messages in the brain.
- However, proteins can sometimes misfold, which in turn leads to harmful and infectious prions.
- If they are not recycled back into the body and continue to misshapen and grow diseased, they can build up in the brain.
- They can also cause other proteins to misfold and clump together.
- That is a Prion Disease, and CJD is actually the most commonly acquired prion disease (even if it is rare)
- This clumping in turn causes brain damage, and as discussed many other symptoms such as memory loss and personality changes. (and it is often fatal, which is an issue as scientists don’t know a lot about prion diseases)
- Prions also have no DNA, because proteins do not have DNA. They are created from messages sent by DNA.
- They duplicate and replicate by infecting other proteins
How do prions specifically cause CJD?
- As mentioned, the prions cause other proteins to misfold as well
- Brain cells will die, and then more prions are created, which attack more brain cells
- When big clusters of brain cells are killed, big clumps of prions called plaques will develop in the brain (and thus further that chain reaction)
- Holes are also created in the brain, making the brain look spongy, which in turn lead to all the symptoms associated with CJD and eventually death.
Causes for each type of CJD
-
Sporadic CJD
-
A protein spontaneously changing into a prion,
-
Variant CJD
-
CJD which is acquired from eating meat infected with BSE or mad cow disease
-
Familial CJD
-
A mutated gene which results in said abnormal prion
-
Iatrogenic CJD - iCJD
-
The spread of CJD through medical instruments
-
Due to regulations, iCJD isn’t nearly as common as it once was.
-
Genetic
-
By genetic mutations which mess up the creation of proteins
Important vocabulary DNA - exists in every cell of your body, and holds your genetic code. The body's instruction manual. There are four chemicals, (adenine) A, (thymine) T, (Guanine) G and (Cytosine) C. These chemicals (nucleotides) write the instructions. RNA- DNA is super important and it makes up all the cells, and therefore can not leave the nucleus. So, RNA is its messenger, going in and out to make things like proteins for the body. Genes- building blocks, and a segment of DNA. Some genes code instructions for the making of proteins, some for instructions for RNA. You get one gene from your mom, and the other from your dad. Chromosomes- these are thread like structures living inside the nucleus of a cell. They contain the DNA, and we have 46 of them, which make 23 pairs *in each cell* The chromosome carries the DNA in the nucleus, which is responsible for building who you are. Further down, genes are segments of DNA which define specific characteristics Genetic mutation- occurs when cells divide and multiply. They are copy and pasting your code, so if something goes wrong and a piece of the code is missing, it could potentially mean a change in how your body functions Amino Acid - Molecules which form to make proteins Protein - an important structure in the human body which provides structure, allows for certain functions, and transports materials. If a protein is mutated, it no longer works, or becomes a prion. PrP^c - is to dictate a prion protein, which are present on neurons and are normal(which are mostly in the brain) PRNP - The gene on chromosome 20, which encodes for the prion protein. The prion protein gene. Prion (PrP^sc) - misfolded prion protein (mutated PrP^c)
What does it do to an individual? // death rates and symptoms
Symptoms The types of symptoms can vary and change depending on the type of CJD
-
Sporadic
-
Affecting the nervous system (neurological symptoms)
-
These symptoms will rapidly worsen with time
-
Variant
-
First psychological symptoms (emotions and behaviour) will develop
-
Then will follow neurological symptoms which will rapidly worsen with time
-
Familial
-
The same pattern as sporadic CJD, however the symptoms will take much longer to process
-
Typically years rather than months
-
Iatrogenic
-
Unpredictable and depends on how the person came into contact with the prion which caused the disease
Neurological symptoms - initial
- Difficulty walking due to problems with balance and coordination
- Speech is often slurred
- A pins and needles feeling
- Feeling dizzy
- Trouble seeing or witnessing hallucinations
Psychological symptoms - initial
- Feeling depressed
- Withdrawal from loved ones
- Feeling anxiety or irritable
- Insomnia or troubles sleeping
Neurological symptoms - advanced
- Ataxia (complete loss of control over a person's body and coordination)
- Muscles twitch and spasm
- Loss of bladder/bowel control
- Becoming blind
- Difficulty to swallow
- inability / loss of speech
Psychological symptoms - advanced
- Severe memory loss
- Concentration issues / feeling confused
- Feeling agitated / aggressive behaviour
- Loss of appetite (leading to weight loss)
- Feeling paranoia / unusual emotional responses
The final stage
- As the disease persists and reaches the final stage, most typically everyone with any version of CJD will become bedridden, and require care at all times
- They are unable to communicate
- Death will soon follow, usually from infections or organ failure
- Nothing can be done at this point to help a patient
Death Rates In Canada
- As reported by the government of Canada, the suspected number of people to have contracted any sort of CJD virus from 1998 to today, is around 2,900, so about 3000 people in total.
- The definitive number of people from this time period to have contracted sporadic CJD is 1381, Iatrogenic six, genetic 99, and variant only two.
- An investigation was conducted by Canadian health professionals and investigators associated with the central CJD surveillance registry (run by the Public Health Agency of Canada), in order to determine cases and mortality rates from CJ
- Said investigation was done through family histories, clinical profiles, and other laboratory investigations. This study was then reported by the National Library of Medicine, and reports the following information following CJD cases from 1998 to 2013. -
- They found 613 cases of sporadic, 43 genetic, 4 iatrogenic, and two variant. Since it is an older study, it makes sense that the numbers don't line up. However, the pattern of sporadic being the most common whilst variant being the least common stays consistent.
- Overall they reported 662 deaths, and reported a 95% confidence interval for their statistics
- However, discussing in a more global sense, according to the CJD foundation 1 to 2 per million per year, which in turn translates to around 500 new cases per year.
- There is one CJD death per every 6000- 10,000 people
- Showing that this disease, while 100% fatal, is very rare
What does it do to the brain?
- As discussed the prions will infect the brain, creating holes and killing brain cells
- As time goes on this will worsen, and therefore symptoms and conditions will worsen as well
- Eventually leading to advanced symptoms, and eventually death
Mentioned above…
- Prions cause other proteins to misfold as well
- Brain cells will die, and then more prions are created, which attack more brain cells
- When big clusters of brain cells are killed, big clumps of prions called plaques will develop in the brain (and thus further that chain reaction)
- Holes are also created in the brain, making the brain look spongy, which in turn lead to all the symptoms associated with CJD and eventually death.
IMPORTANT VOCABULARY Psychological - to do with the mind / brain - an emotional state of being Neurological- to do with the nervous system Nervous System - a network of nerves and fibres which sends messages from the body to the brain and vice versa.
What individuals are affected?
- There may be a risk of contracting CJD if an individual has family history with prion diseases, has eaten meat infected with BSE, or came into contact with medical equipment infected by CJD
- Most often seniors are affected by CJD, typically between the ages of 45 and 75.
- Most often CJD is diagnosed as sporadic (85%), so the reasoning is often unclear as to why the disease developed (why prions had developed.) But it is speculated that this prion disease is more common amongst older people because they are more likely to have their proteins spontaneously fold as compared to younger individuals.
- However it is important to keep in mind that the disease is still very rare, even if it is more common amongst an older population
- And variant, genetic, and iatrogenic versions of CJD are still possibilities, however even more so unlikely
Why is it so far uncured?
- Prion diseases are irreversible, unable to be overcome by the immune system
- As prions are seen by the body as proteins, and does not recognize it as a foreign body like a bacteria. Therefore allowing said disease to develop further (in its spontaneous, quickly clumping, and deadly nature. This clumping is known as FIBRILS)
- And on top of that, there is no medication or drugs for the treatment of prion disease. This is speculated to be the case due to the fact that there isn’t enough attention or research done on the disease, because it is so rare.
- Prions, unlike bacteria and viruses, have no nucleic acid genetic element (DNA or RNA), and therefore can not be destroyed by ultraviolet irradiation (exposing to radiation) as that is what would kill said nucleic acid.
- They also resist protease, an enzyme which degrades proteins. Making prevention even more difficult
- Due to an unclear understanding of what prion proteins do for the brain, it is unclear how to handle treatment because there is a risk of disturbing something important (these prion proteins are in the brain after all, one of the most crucial organs)
Important vocabulary Fibrils - insoluble clumps of prions in the brain
What have hospitals and scientists been doing in the meantime? // diagnoses, treatment, prevention
How is CJD (and prion diseases) diagnosed? Prion diseases are confirmed by taking a sample of the brain during a biopsy. (or after the death of a patient) However, a biopsy is risky, as it's taking the tissue of the brain out of someone who is living. So health care professionals instead run many other tests to detect a prion disease. These include…
- MRI (magnetic resonance imaging) scans of the brain. By taking pictures of the brain with radio waves, a doctor is able to see that spongy texture of the brain which often results from CJD. (MRI’s result in high quality and clear images, and are often used for these kinds of purposes)
- Spinal Tap (lumbar puncture) is when you take samples of fluid from the spinal cord or around your brain. When such a fluid is extracted, it can help eliminate other diseases that have similar symptoms to CJD and helps to see the levels of proteins, which can then point to the possibility of a prion disease.
- Cerebrospinal Fluid (CSF ) Tests is a branch of tests with spinal taps fall under. The test looks for elevated markings of proteins which dictate rapid brain cell death, which is a common factor of CJD. There is a newer test based in CSF called RT-QuIC which can actually detect certain prions which cause CJD.
- Electroencephalogram (EEG) which is a test done to look at brain waves with the use of electrodes. This is a method which measures the brain's electrical activity. If an individual has CJD, it will show an abnormal pattern.
- Blood tests
- Visual and Neurological exams to check for symptoms (nerve damage, vision loss)
- Genetic tests for the potential a prion disease is hereditary
How are Prion Diseases Treated?
- Currently, there is no cure for prion diseases. Health care professionals try to make sure that a patient with prion disease is safe and comfortable even in their final moments
- Researches have however been searching for drugs that could control CJD, and other treatments (which I’ll research when I want to see what I can do)
- There is the CJD foundation, which is an entire organization based around providing support in the form of funds, awareness, and healthcare for families and individuals affected with CJD. They also allow for research to be submitted, in order to progress knowledge on CJD and prion disease as a whole.
14-3-3 PROTEINS
- As we know, proteins play a big role in all of our bodily and cell functions from structure, development, survival, growth, and death
- The family of 14-3-3 proteins are highly conservative. This means that members of said family are very similar in their structure.
- They are found in all eukaryotic cells
- They serve as a ADAPTER CHAPERONE MOLECULE, (a type of accessory protein that prevents misfolding whilst also moving) and help in cellular functions by moving from the cytoplasm to the nucleus of a cell (and vice versa)
- Humans have seven different versions or ISOFORMS of the 14-3-3 protein
- But how do they relate to neurological disorders?
- Under normal conditions, a 14-3-3- protein (like 14-3-3 sigma) performs its usual function (in this case, serving as a blockage for any tumors)
- However, if there is a problem, that protein becomes harmful (say for example the protein 14-3-3 gamma, acting as an oncogene and is able to create cancer, rather than suppress it)
- When the gene is normal, its normal function takes place. When there is a mutation, an oncogene is activated, increases that person's risk to disease such as cancer
- The more mutations (and losses) lead to a higher chance of that patient obtaining that disease (such as cancer)
- Many factors can affect why a gene mutation includes external factors, age, and genetics.
- 14-3-3 proteins are molecules which have remained unchanged throughout evolution, (Conserved regulatory molecules) showing that they are extremely important for biological processes (such as gene expression, signal transduction, and cellular metabolism. They live within within eukaryotic cells (cells with a nucleus)
- They bind with SIGNALING PROTEINS such as kinases, phosphatases, and transmembrane receptors
- Signaling proteins are important because they act as messengers to coordinate activities like growth and immunity. They trigger cellular responses from signals received by hormones and neurotransmitters to transfer information within cells and between cells.
- Both kinases and phosphates communicate and control a cell's behavior, hence what they are signaling proteins.
- This function can allow 14-3-3 proteins to play a role in a wide range of vital regulatory processes (Important functions; allow cells to stay healthy (regulate activities like growth, metabolism, and cell survival.)
- In summary…The protein 14-3-3 can bind those specific signaling proteins together, which can assist in processes which keep organisms healthy and alive.
- 14-3-3 proteins are found in the brain, and are associated with the brain's function as well as disorders
- It is expressed in the neurons of cells, and are produced during the brain's development (this implies it has significance in the brain's development)
- The expression of this protein as per various brain disorders (such as CJD) also shows an impact on the brains plasticity (that being the brain's ability to form new connections in its structure as per external responses such as learning and injury)
- Involved in the making of neurotransmitters (as they activate tyrosine and tryptophan hydroxylases, which make said neurotransmitters)
- They are involved in many cell functions! (including including cell survival, growth, differentiation, migra-tion, and signaling)
- They are able to bind (connect) with some intracellular (inside the cell) proteins such as transmembrane receptors, cytoskeletal proteins, and signal-transducing proteins, such as kinases and phosphatases
- This function allows for the following to happen; the regulation of transcription, cell-cycle control, protein trafficking, metabolism, signal transduction, stress response, and apoptosis
HOW DO THEY RELATE TO CJD?
- Creutzfeldt-Jakob Disease (CJD) is a fatal degenerative brain disorder caused by a misfolded protein, PrPSc prion (scrapie isoform of the prion protein), with the symptoms of confusion, depression, abnormal body sensations, autonomic nervous sys-tem disorders, and dementia (directly from website)
- 14-3-3 proteins have been thought to serve as a biomarker for the diagnosis of CJD
- Finding 14-3-3 proteins in cerebrospinal fluid was often seen as an indication of CJD, concluding that its presence serves as a useful diagnostic tool for the disease
- However, as always, things change…
- In other neurological diseases, (Rasmussen’s encephalitis, Schilder’s disease, or diffuse large B-cell lymphoma), the 14-3-3 protein was not found
- This may then mean that the presence of 14-3-3 proteins ISN'T because of cell death causing it to leak everywhere, but rather the way in which the proteins actually fold within the brain
- While all isomers of 14-3-3 were found, two were found in specific; 14-3-3 beta and gamma, more specifically now 14-3-3 gamma, be used as a DETECTION for CJD.
- 14-3-3 proteins are found in abundance within the neuron. When proteins mutate, and prions form, creating spongiform degeneration will those proteins be released
- An antibody of a 14-3-3 protein reacted with cerebrospinal fluid (which is a clear fluid around the brain and spinal cord which serves protection)
- Patients with CJD had this 14-3-3 protein.
- Meanwhile healthy individuals or individuals with other diseases such as Alzheimer's did not have this protein.
- This helps us differentiate between these two diseases (CJD and Alzheimer's) which have very similar symptoms. Therefore, allowing proper treatment as needed in order to maximize a patient's health before symptoms arise.
PENTOSAN POLYSULFATE
- Pentosan polysulfate (PPS) is a commonly used medication to treat bladder pain, relieving the symptoms of pain and discomfort that come with it.
- However, Pentosan polysulfate (PPS) has shown some results in mice, prolonging the incubation periods those infected with prion diseases
- PPS seemingly prolongs the lives of patients who take it, however does not halt the neurodegeneration done by the prions
- While usually taken in pill form for bladder pain, it in injected directly into the brain of patients with CJD
- There are apparently four possible situations
- The estimate for the expected survival of a patient is wrong, as previous patients may have been diagnosed late in the course of their disease.
- These patients survived longer due to aggressive treatments for other conditions (like pneumonia) (however data suggests this isn't a likely case)
- It is a simple chance finding due to the very small sample
- PPS actually works in treating CJD!
- Ian Bone says the following about the study conducted
- “There are some things the families can take away from this. There will now be more research, particularly animal research, because we need to measure the drug’s penetration into the brain. Also, the concerns over possible dangerous side effects seem to be groundless. And there is limited evidence of prolongation of life.”
RT-QuIC
- Real time quaking induced conversion or RT-QuIC, has been a way for clinical diagnoses of CJD
- transmissible spongiform encephalopathies are often a sure fire sign or PrPsC, so detecting it is often the first logical step
- This test does just that! By taking a fluorescent die called thioflavin T, is able to bind to aggregates proteins
- CSF (cerebrospinal fluid) is extracted, and mixed with rPrP (Recombinant Prion Protein, which is used for research)
- If misfolded prions are detected they bind to the rPrP, causing the formation of long fibrils, indicating the presence of disease
- The time between the creation of the fibrils is called the lag phase (it can last up to 30 hours!)
- Once they do this, they bind to the dye, which lights up under certain lighting
- This allows for real time tracking of the diseases progression
- The rate of which the aggregation occurs follows a sigmoidal curve,
- This curve indicates that initially, there's little to no aggregation, then it accelerates, and finally, it levels out as the system reaches a maximum point of aggregation (clumping)
- Sensitivities (test those with the disease) 69%-89%
- Specificity (test those without the disease) 99%-100%
- The figure shows the RT-QuIC reactions // ThT, thioflavin T. detected as per sporadic Creutzfeldt-Jakob disease brain homogenate as positive control (red), Alzheimer’s disease brain homogenate (yellow). This shows CJD detects high prion aggregation
- This one shows the “lag phase” of prion aggregation as per sporadic CJD (red) and another unrelated prion disorder (blue)
- Despite high performance and accuracy, the process is very long (taking up to 90 hours, so about 3.75 days). An improved version of RT-QuIC called RT-QuIC (IQ) uses hamster rPrP as a substrate, providing similar results but much less observational time (30 hours)
PRIONS IN YEAST
IMPORTANT VOCABULARY Self propagating- induce and cause other proteins to change shape Alternative confirmations- the different shapes a protein can take; important due to the different functions different conformations can entail. Primary sequence- the chain/order of amino acids. Yeast and human prions are different in their structure Yeast Saccharomyces Cerevisiae- commonly known as bakers/brewers yeast. ------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------
- Prions are not only found in animals, where they can cause extremely harmful neurodegenerative diseases; they can also be found in fungi such as yeast (Saccharomyces cerevisiae)
- Like human prions, these fungal prions are also able to change their conformation on their own terms. Adopting multiple shapes/conformations, and causing other proteins to do the same. (they are able to exist in one or more self-propagating alternative conformations)
- However, continuing this comparison, they share a little primary sequence relationship with human/mammal prion proteins.
- Fungal prions hold a great diversity of proteins, which do participate in key cellular mechanism (such as transcription and translation)
- “Upon switching to their prion form, these proteins can generate stable, sometimes beneficial, changes in the host cell phenotype.”
- This direct quote from Pubmed means that when a protein is stable, it can maintain its shape for a good while (allowing self propagation, or the transfer of changes to other cells)
- However, unlike in mammals, these misfolded prions can provide unforeseen benefits to the host, such as how it grows and adapts (the phenotypes)
IMPORTANT VOCABULARY Fragmentation- the process of being broken down into smaller parts Disaggregation- The opposite of aggregate; to break into smaller parts, and then analyzing those individual components of the original aggregate Exogenous- relating to external factors; something which comes into an organism from the outside Important topic-chaperones
- Hsp104 is a chaperone (which is defined as a protein which helps the other proteins) specifically to fungi such as yeast. This chaperone is interesting, because rather than preventing the misfolding of proteins like chaperones Hsp40, 70, and 110, by working together, sharing information, etc, it takes the already aggregated proteins due to stress such as heat, toxins, etc, and helps it to unfold and keep the fungi safe.
- As we know, amyloids are a certain type of protein aggregate characterized by its high beta sheet content, and fibral like structure. These types of amyloid aggregates are found in animals, they are similar to the prions found in yeast and fungi as well.
- The propagation, or reproduction/spreading, of said fungal prion amyloids can be done through the chaperone Hsp104
- Now this chaperone usually works to unfold infected prions, if it does so too small it may actually lead to the prions misfolding even more as more normal proteins become infected
- Animals such as humans do not have this certain chaperone, but we do have others such as Hsp 40, 70, and 110 which all help with the disaggregation of prions by stopping them from misfolding in the first place
- If Hsp104 is introduced (exogenous Hsp104) from another source, it may be able to team up with these chaperones and promote disaggregation in humans and therefore help with treating diseases associated with amyloids
- But there is still doubt that this would work at all, as scientists worry that Hsp104 alone may not be enough. These limitations may mean that other treatments will have to be used alongside this introduction of a fungal chaperone.
- The study was to show and explain findings on how certain chaperones in fungi and humans can work together, and then perhaps lead to certain therapeutic treatments for certain diseases associated with them (amyloid diseases, such as CJD)
IMPORTANT VOCABULARY Self propagating- a system which can continue its existence, without needing external energy/ influence to sustain it. Biological properties- a characteristic of features of a living organism, relating to its structure/function/behaviour etc. Chaperone imbalances- an imbalance within the normal levels of chaperones (assistants) within a cell/ organism
CLARIFICATIONS Self propagating vs. Self Templating- A misfolded protein in a system can self-template, meaning it can use itself as a guide to let other proteins misfold in the same way. This at the same time is self propagation, because the misfolded proteins are able to sustain themselves by themselves using self templating Chaperone imbalances As we know, chaperones are helpers. They can assist either proteins OR prions. When there is an imbalance among the chaperones can lead to a variety of issues such as misfolding/aggregation.
RECAP
- Yeast prions have become important models for human prion and amyloid diseases.
- A prion protein can misfold into many different amyloid forms. These different forms have different characteristics and are called protein VARIANTS or STRAINS.
- These prions are self propagating and have different biological properties.
- The beta sheet structure within yeast prions is suggested to be the reason prion variants can pass along their information (self templating)
------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------
- As we know, chaperones exist to help both the proper folding of proteins, but can also help prions thrive
- Because of this, said yeast prions can propagate with these chaperones
- If there is an IMBALANCE, then it may stop the spreading of yeast prions and cure it
- Btn2/Cur1 can help heal the prion strain URE3. These two proteins recognise misfolded prions, and target them to prevent their accumulation, and therefore their spread. They do this through DEGRADING the protein, or INHIBIT THE PROPAGATION.
- The article mentions two prion strains found in fungus- PSI+ and URE3
- PSI+ is a variant which affects the translation of proteins, which can lead to non-functional proteins
- While it can be lethal if in large amounts, however it is often mild and can allow the yeast to handle stressful conditions.
- The other strain URE3 is related to nitrogen metabolism, and like PSI+, depending on the quantity it can either be lethal of helpful
- The study suggests that because these variants are rare, they could have severe detriments (such as toxicity or decrease in reproduction) to yeast despite their beneficiary properties.
- However, in a surprising twist, the protein variant [Het-s] is beneficial to the host.
- It participates in something called heterokaryon incompatibility which can help an organism distinguish its cells from different ones with a different genetic background, preventing the spread of harmful genetic elements in that organism
- This difference highlights the complexity of the fungal protein structure.
QUESTION
Can the protein Btn2 and Cur1 be used exogenously like the chaperone protein Hsp104 to cease the aggregation of the prion protein, therefore stopping or slowing neurodegeneration?
HYPOTHESIS
If Btn2 and Cur1 proteins are introduced exogenously into the human brain from saccharomyces cerevisiae then will the aggregation of mammalian prions in the human brain slow because Btn2 and Cur1 helps degrade and inhibit the propagation of the prion strain URE3 found in bakers yeast.
Btn2 and Cur1 PROTEINS
- URE3 prion is the mutated form of Ure2 (performs nitrogen catabolism) in yeast
- Btn2 and Cur1p can cure this prion when overexpressed
- Cur1 is a paralog or Btn2, meaning its from the same species from the same ancestor and formed from gene duplication
- Btn2 colocalizes (is present at the same time as) Ure2 aggregates
- URE3 prions will get larger in size if Btn2 and Cur1 aren’t present - it can be cured when the two proteins are back to at least a normal amount
- Btn2 and Cur1 are members of the hook protein family
- They are metazoans (members of kingdom animalia as they are multicellular heterotrophic eukaryotes) which use microtubules (small hollow protein tubes in a cell) to move things like organelles or even aggregates in them
- Those organelles and aggregates are the cargo which are moved by the motor (motor proteins) kinesins and dyneins with ATP. The microtubules are the tracks
- The hook proteins Btn2 and Cur1 HOOK the motors and cargo together
How do Btn2 and Cur1 work?
- Sequestration is how Btn2 and Cur1 work
- They gather the prion aggregates in a single location, prevent their division to daughter cells, and thus there is no more prion aggregation because it is no longer being passed down!
- This is suggested/what is assumed to be happening as Btn2 is colocalizes (is present at the same time as) Ure2 aggregates
- Both proteins are needed for efficient curing
------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------
BIOLOGICALS - medicines which are derived from living organisms such as plants, animals, cells, etc than making them artificially with chemicals
- 7/10 of the top selling drugs are made of proteins (biologicals)
- Their large size, structural complexity, and molecular diversity often results in them being able to recognize receptors that challenge and evade (basically harm) smaller molecules
- However! There is an issue. Most proteins are not actually able to get through the phospholipid bilayers of these mammalian cells!
- The limitation has prevented disease causing and relating receptors that proteins would have been able to deal with
- Because of this, there is a lot of work being done to get over this hurdle.
- Often through the exogenous proteins to be delivered to cells (intracellular delivery) in mammalian (human) cells
- These can be natural or engineered
------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------
- There are several methods which transport nucleic acids (DNA or RNA, which are able to make/encode proteins) into cells
- CATIONIC LIPIDS which are positively charged molecules that make liposomes to carry said nucleic acids
- VIRUSES as they can infiltrate cell walls naturally (probably wouldn't want to use that…)
- Breaking open the cell membrane to get whats needed inside the cell
- CELL-PENETRATING PEPTIDES (CPPS) are short peptide chains that help proteins enter cells (sometimes to specific cells)
- PROTEIN RESURFACING changes the surface of the proteins used to allow them to fit in better
- Using proteins from TOXINS as they can naturally penetrate the cell barriers
- CHEMICALLY MODIFIED PROTEINS which change the shape with chemicals to allow them to better fit
- NANOPARTICLES which surround and capture proteins within them like a capsule to help transport them better
The question remains…would it be better to exogenously introduce proteins into the brain through nucleic acids that encode for proteins or directly inserting the protein itself? Well, as we know, the brain is a very delicate organ. If we inserted new DNA to the brain, how would that itself impact the brain? Would it cause more complications than simply inserting the protein as is? I think inserting the protein by itself contains less risk with the same affect. Of the many ways the article lists on how to insert proteins, CPPs seem the least invasive or risky.
- Directly inserting the proteins is a far simpler way to introduce exogenous proteins to mammalian cells
- But with the hydrophilic surface of the cells, protein transfers are limited
- With the rise of biologics in the pharmaceutical industry, many methods have been uncovered in order to insert exogenous proteins into mammalian cells
Cell-Penetrating Peptides (CPP’s)
- Relatively short peptides (less than 20) that can attach to a protein and help it get over the phospholipid bilayer of a cell.
- Very straightforward for direct delivery of the protein to the cell.
- But between common examples of cpp’s, shows a common trend of high cationic residues (positively charged amino acids) in particular lysine and arginine, two types of amino acids. (this includes Tat peptide from the trans-activating protein Tat of HIV-1, and the third helix of the homeodomain of antennapedia called penetratin)
- Cationic residues react in nice ways when met with the phospholipid bilayer of a mammalian cell by engaging with negatively charged glycosaminoglycans (GAGs)such as heparin sulfate and chondroitin sulfate on the surface of cells. Specialized glycoproteins (class of proteins where carbohydrates groups are attached to the peptide chain) called proteoglycans, have a core protein which is attached to the GAGs.
- glycosaminoglycans are long, negatively charged polysaccharide found on a cells surface.
- chondroitin sulfate and heparin sulfate is a sulfated GAG.
- They have functions in regulating cell growth, proliferation (rapidly protruding parts of a cell), promotes cell adhesion, anticoagulation, and wound repair
- Polypeptides with more arginine are better at transportation than lysine
- They react with the proteoglycans through bidentate interactions. This means the bond happens in two places, making it stronger (and the transportation better)
- The cell will then engulf and internalize those cells (endocytosis)
- A limitation that exists however, is that these polycationic CPPs like to bind and enter really any mammalian cells. This means that the cells (in our case, the protein cells in the brain) may not get targeted like we want
- Because of this, the cargo they carry can go to undesirable cells which could lead to unwanted side affects
- An example of how CPPs has been made more specific includes cancer-cell activates CPPs
- These cells have three components;
Polycationic CPP to penetrate the cell membrane Polyanionic biopolymer- binds to the polycationic CPP to stop it at a certain point from entering healthy cells
- The polyanionic biopolymer binds to the cationic CPP and stays linked when no cancer associates proteases are present. This keeps the cpps from penetrating any cells including healthy ones
- But when cancer is detected, and it releases a protease, which disconnects the link between the cationic and anionic parts of the cpps. And they separate, the cationic cells able to penetrate cell membranes
- This means the cationic cells are able to be targeted specifically by cancel cells and act upon them only
The protein BTN2 has cationic parts to it, but is not necessarily considered a high cationic protein.
- However, the thought process and research continues. It has been thought possible to mutate some proteins to add or replace amino acids with cationic residues to increase their positive charge, which increases the ability to bond with negatively charges aspects of the cell membrane, making the bond stronger
- When arginine is used as a replacement for amino acids, it is called arginine grafting.
- Take for example RNase, an enzyme which can destroy RNA found within cells. When it is modified with the cation arginine, they can enter the cells and can cause the death of said cell.
- Something to keep in mind however, is the fact that the proteins may get trapped in small compartments within the cells called endosomes. This could prevent the movement of the proteins, meaning they don't act to their full capacity
- Scientists are still trying to navigate around this obstacle to more desirable areas of a cell such as the nucleus.
Supercharged proteins
- These are proteins modified to have a lot of cations
- The main benefit they have is that they not only can penetrate mammalian cells, but are stable and therefore won’t aggregate or fold
---------------------------------------------------------------------------------------------------------------------
- Most proteins actually aren't able to go through this type of genetic mutation
- So instead, scientists have been looking into using NANOBODIES
- Nanobodies are proteins derived from camels and llamas that have structures that allow them to bond to other molecules due to their unique beta-sheet structure and CDRs (complementary determining regions)
- Cationic resurfacing can be done to these nanobodies, and can then effectively enter the mammalian cells
Data
Figure #1
This diagram illustrates how cell penetrating peptides interact will cells.
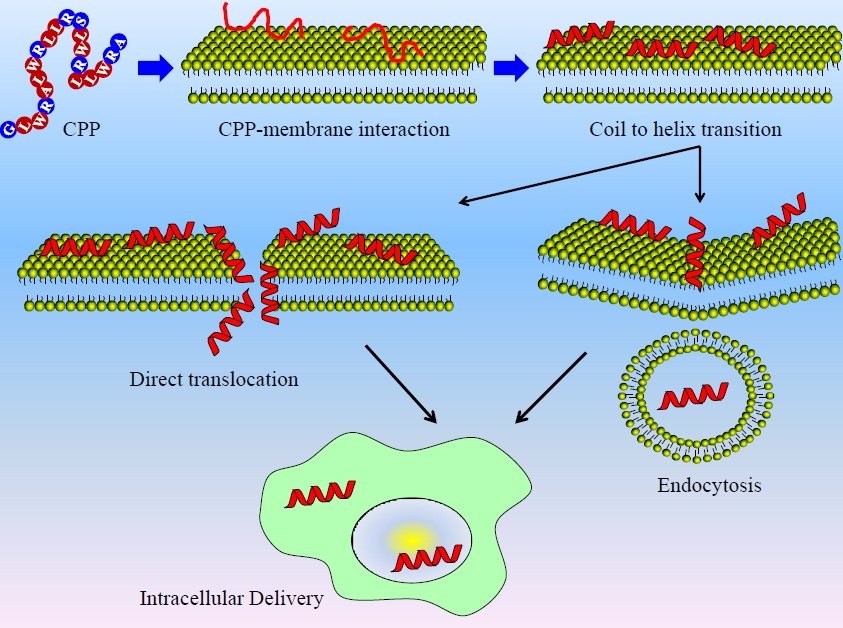
Figure #2
This diagram illustrates the mechanisms around selective CPPs

Conclusion
CONCLUSION
Btn2 and Cur1 proteins are able to silence misfolded proteins that can form within saccharomyces cerevisiae by gathering the prion aggregates in a single location. Btn2 and Cur1 prevent aggregated proteins from dividing into daughter cells, which stops their traits from being passed down, ceasing aggregation within the yeast. This sequestering done by the proteins can help mammalian cells fight off prion diseases as well, however, Btn2 and Cur1 proteins are not present within them. But using the practices of biologics, it is possible to extract Btn2 and Cur1 proteins from saccharomyces cerevisiae, and insert them into mammalian cells within the human brain exogenously. This would sequester those proteins within the human brain the same way it does in the fungi.
Cell penetrating peptides are one of the simplest ways to introduce exogenous proteins such as Btn2 and Cur1 into mammalian cells. These small peptides attach to the desired protein, assisting in its transportation. The cationic residues on CPP’s (such as arginine and lysine) bind to the phospholipid bilayer of a mammalian cell by engaging with negatively charged glycosaminoglycans (GAGs) such as heparin sulfate and chondroitin sulfate on the surface of cells. Through this bond, the protein which is attached gets engulfed by the cell through endocytosis. From this point, the Btn2 and Cur1 proteins are expected to act as they usually would within yeast cells, and sequester the protein aggregates.
LIMITATIONS
A limitation that exists however, is that these polycationic CPPs like to bind and enter most mammalian cells. This means that the cells we want the protein to bind to, may not get targeted like we want because they are so general. Because of this, the cargo they carry can go to undesirable cells, which could lead to unwanted side effects.
Efforts have been made to make CPPs more specific. An example of this includes cancer-cell activated CPPs. These cells have a few components; Polycationic CPP to penetrate the cell membrane, and polyanionic biopolymer, which binds to the polycationic CPP to stop it from entering healthy cells. The polyanionic biopolymer binds to the cationic CPP and stays linked when no cancer associated proteases are present. This keeps the CPPs from penetrating any cells including healthy ones. But when cancer is detected, it releases a protease, which disconnects the link between the cationic and anionic parts of the CPPs. And they separate, the cationic cells able to penetrate cell membranes. Meaning the cationic cells are able to be targeted specifically by cancer cells and act upon them only.
However, what would happen if the Btn2 and Cur1 proteins aren't able to go through this genetic mutation? Scientists have navigated around this obstacle as well, and have landed on using nanobodies. Nanobodies are proteins derived from camels and llamas that have structures that allow them to bond to other molecules due to their unique beta-sheet structure and CDRs (complementary determining regions) Cationic resurfacing can be done to these nanobodies, and can effectively enter the mammalian cells
FUTURE STUDY
Applying what we know about cancer-activates CPPS, it may be possible to apply that same knowledge to prion diseases. When misfolding is occurring within the brain, then will the Btn2 and Cur1 proteins be released in order to sequester them. That way, healthy proteins are not being targeted. Diagnosing CJD within patients also becomes less strenuous because if the Btn2 and Cur1 proteins are inserted and work like cancer activates CPPs, they will handle them accordingly rather than festering into a larger issue.
However the doubt remains on how the brain will react to foreign entities inside of it, such as these Btn2/Cur1 proteins, as well as potentially nanobodies. Will the brain/immune system attack these foreign entities? How are we supposed to infer the potential risks of biologics, especially to something as delicate as the human brain? These questions raise the possibilities for future studies and works to be conducted upon prion disease and exogenous proteins.
Citations
Mayo Clinic. (2023, January 28). Creutzfeldt-Jakob disease. Mayo Clinic. https://www.mayoclinic.org/diseases-conditions/creutzfeldt-jakob-disease/symptoms-causes/syc-20371226
National Institute of Neurological Disorders and Stroke. (2023, January 23). Creutzfeldt-Jakob Disease. Www.ninds.nih.gov. https://www.ninds.nih.gov/health-information/disorders/creutzfeldt-jakob-disease
NHS. (2021, September 7). Creutzfeldt-Jakob disease. NHS. https://www.nhs.uk/conditions/creutzfeldt-jakob-disease-cjd/
John Hopkins Medicine. (2025). Prion Diseases. Johns Hopkins Medicine. https://www.hopkinsmedicine.org/health/conditions-and-diseases/prion-diseases
NHS Choices. (2019). Causes - Creutzfeldt-Jakob disease. NHS. https://www.nhs.uk/conditions/creutzfeldt-jakob-disease-cjd/causes/
Mayo Clinic. (2023, January 28). Creutzfeldt-Jakob disease. Mayo Clinic. https://www.mayoclinic.org/diseases-conditions/creutzfeldt-jakob-disease/symptoms-causes/syc-20371226
Cleveland Clinic. (2022, May 20). DNA, Genes & Chromosomes Overview. Cleveland Clinic. https://my.clevelandclinic.org/health/body/23064-dna-genes--chromosomes
NHS. (2019). Symptoms - Creutzfeldt-Jakob disease. NHS. https://www.nhs.uk/conditions/creutzfeldt-jakob-disease-cjd/symptoms/
John Hopkins Medicine. (2025). Prion Diseases. Johns Hopkins Medicine. https://www.hopkinsmedicine.org/health/conditions-and-diseases/prion-diseases Canada, P. H. A. of. (2009, June 6). Creutzfeldt-Jakob disease surveillance system report. Aem. https://www.canada.ca/en/public- health/services/surveillance/blood-safety-contribution-program/creutzfeldt-jakob-disease/cjd-surveillance-system.html
Coulthart, M., Jansen, G., Connolly, T., D’Amour, R., Kruse, J., Lynch, J., Sabourin, S., Wang, Z., Giulivi, A., Ricketts, M., & Cashman, N. (2015). Creutzfeldt-Jakob disease mortality in Canada, 1998 to 2013. Canada Communicable Disease Report, 41(8), 182–191. https://doi.org/10.14745/ccdr.v41i08a01
CDC. (2024, April 22). About Prion Diseases. Prion Diseases; CDC. https://www.cdc.gov/prions/about/index.html
The Latest in Medicine: The Problem of Prions Disease. (n.d.). Minds Underground. https://www.mindsunderground.com/muarticles/prions-disease
Aucouturier, P., Carp, R. I., Carnaud, C., & Wisniewski, T. (2000). Prion Diseases and the Immune System. Clinical Immunology, 96(2), 79–85. https://doi.org/10.1006/clim.2000.4875
Creutzfeldt-Jakob Disease Foundation. (n.d.). Creutzfeldt-Jakob Disease Foundation. https://cjdfoundation.org/
National Institute of Neurological Disorders and Stroke. (2023, January 23). Creutzfeldt-Jakob Disease. Www.ninds.nih.gov. https://www.ninds.nih.gov/health-information/disorders/creutzfeldt-jakob-disease
Mayo Clinic. (2018). Creutzfeldt-Jakob disease - Diagnosis and treatment - Mayo Clinic. Mayoclinic.org. https://www.mayoclinic.org/diseases-conditions/creutzfeldt-jakob-disease/diagnosis-treatment/drc-20371230
Cho, E., & Jae Yong Park. (2020). Emerging roles of 14-3-3γ in the brain disorder. Journal of Biochemistry and Molecular Biology, 53(10), 500–511. https://doi.org/10.5483/bmbrep.2020.53.10.158
Samantha Churovich. (2026). 14-3-3 Proteins. Youtu.be https://youtu.be/FOAtUaS3inE
Green, A. J. E. (2018). RT-QuIC: a new test for sporadic CJD. Practical Neurology, 19(1), 49–55. https://doi.org/10.1136/practneurol-2018-001935
Tuite, M. F., Marchante, R., & Kushnirov, V. (2011). Fungal prions: structure, function and propagation. Topics in Current Chemistry, 305, 257–298. https://doi.org/10.1007/128_2011_172
Glover, J. R., & Lindquist, S. (1998). Hsp104, Hsp70, and Hsp40. Cell, 94(1), 73–82. https://doi.org/10.1016/s0092-8674(00)81223-4
Wickner, R. B., Edskes, H. K., Gorkovskiy, A., Bezsonov, E. E., & Stroobant, E. E. (2016). Yeast and Fungal Prions. 191–236. https://doi.org/10.1016/bs.adgen.2015.12.003
Wickner, R. B., Bezsonov, E., & Bateman, D. A. (2014). Normal levels of the antiprion proteins Btn2 and Cur1 cure most newly formed [URE3] prion variants. Proceedings of the National Academy of Sciences, 111(26). https://doi.org/10.1073/pnas.1409582111
Bruce, V. J., & McNaughton, B. R. (2017). Inside Job: Methods for Delivering Proteins to the Interior of Mammalian Cells. Cell Chemical Biology, 24(8), 924–934. https://doi.org/10.1016/j.chembiol.2017.06.014
Acknowledgement
All of my thanks goes towards Ms.Fan! I would like to thank her for her insight, encouragement, and assistance with this project, and I'm very grateful to have had someone as cool and smart help me throughout the entire process.