
Design and Construction of CRISPR/Cas9-Mediated Knockout Plasmids for Autism-Risk Genes
Jingyi Xiang
Grade 11
Presentation
Hypothesis
1. By optimizing the backbone plasmid as well as the molar ratio of the backbone to inserts, knockout plasmids for 10 ASD risk genes can be successfully constructed.
2. An increase in the molar ratio of the backbone plasmid to the gRNA oligo insertions would positively correlate with the proliferation of clones.
Research
Autism Spectrum Disorder (ASD) is a neurodevelopmental disorder that affect approximately 1 in 44 children.1 Autism typically manifests in early-onset difficulties in social communications and repetitive and restricted interests and behaviors.2 According to the fifth edition of the American Psychiatric Association’s Diagnostic and Statistical Manual, a child must present with such characteristics that cannot be explained by other neurodevelopmental disorders in order to be diagnosed as autistic, or else they would be diagnosed with other disorders or considered comorbid.3 In addition, it should be mentioned that the male to female ratio for ASD is between 3:1 and 4:1.4 This along with family and twin studies gave rise to the female protective effect hypothesis, which states that females may need more genetic mutation than males to express the autistic phenotype. Although environmental factors such as prenatal exposure to pollutants could contribute to the development of ASD, twin studies have shown strong evidence that it is predominantly genetic factors that lead to ASD.5,6
In fact, ASD is one of the most highly heritable disorders and it is known for its heterogeneity.2,7 Approximately 400-1000 autism-risk genes have been identified and have been found to be associated with the formation of synapses, regulation of transcription, and the formation of chromatin.8
To study the effects of autism-risk genes in cell and animal models, efficient tools to mediate the loss of function for specific genes are essential. Cre-lox, FLP-FRT, CRISPR/Cas9 systems are commonly used methods to approach gene knockout.9 Compared to Cre-lox and FLP-FRT, CRISPR/Cas9 is of lower cost and higher efficiency and had thus been widely used for inducing autism-risk gene knockouts.9 For instance, variants of the KCTD13 gene are known to be associated with various neuropsychiatric disorders, including ASD.10 KCTD13 was knocked out using CRISPR/Cas9 in human-induced pluripotent stem cells, and the outcomes pointed researchers towards investigating the ERBB pathway as a potential therapeutic target for disorders associated with KCTD13 deficiency.10 Another study involving the CRISPR/Cas9 mediated knockout of oxytocin receptor genes in prairie vole model demonstrated the crucial role of the gene in regulating behavior.11
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/ CRISPR-associated protein 9 (Cas9) is a gene-editing technique allowing for up to 500 kilobase of DNA segment knockout.12 Originally developed by bacteria to provide acquired immunity against viruses, CRISPR/Cas9 is now used for genetic editing.13
In order to introduce deletions, the ultimate goal of CRISPR/Cas9 is to induce a double strand-break (DSB) in the target gene sequence and activate the non-homologous end joining pathway (NHEJ) in the host genome.14 First, a single guide RNA (gRNA) which contains the custom-designed short CRISPR RNA (crRNA) fused to trans-activating crRNA (tracrRNA) must be synthesized.13 The crRNA contains the spacer, which is identical to the genetic sequence that will be modified and the tracrRNA contains sequences complementary to those downstream of the spacer. This structure allows for the crRNA to bind to tracrRNA, and together with the Cas9, they will form the CRISPR effector complex.13 The most commonly used Cas9 is derived from Streptococcus pyogenes and it can recognize and target a 20 nucleotides-long DNA sequence as well as the protospacer adjacent motif (PAM) 5’-NGG-3’.15 It should be noted that the function of PAM is critical, since it distinguishes the protospacer, which is located in the target genome, from the spacer in crRNA. As a result, a DSB in the specific location of the genome can be introduced by the CRISPR effector complex.
Following the occurrence of a DSB, the NHEJ pathway is utilized by the cell DNA repair mechanisms in the target genome in the absence of a repair template. In brief, the ring-shaped Ku70/Ku80 protein heterodimer binds to the two ends of DSB; the protein then recruits the DNA-dependent protein kinase catalytic subunit, which in turn gathers the DNA ligase complex LigIV-XRCC4-XLF that ligates the DSB ends.16 NHEJ may create insertions, deletions, or substitutions in the target gene, which all ultimately render the gene non-functional.
CRISPR/Cas9 has been widely adapted for gene knockout in cells and animal models.15 However, in many cases the use of one gRNA has proven to be inefficient.17 To design knockout plasmids with two gRNAs targeting the same gene is a strategy to improve the efficiency of gene knockout.17 To reach this goal, the aim of this study is to incorporate two U6-driven gRNA expression cassettes in one plasmid as a basis to construct the knockout plasmids. This kind of plasmid contains a fluorescent reporter expression cassette, two U6 promoter driven gRNA expression cassettes. When the plasmids are introduced into cells expressing Cas9 protein, cells are labeled with fluorescent protein and certain genes are knocked out at a high efficiency by two designed gRNAs. In this project, 10 autism risk genes that are insufficiently investigated are chosen as targets. These knockout plasmids can be used in cell and animal models to study the foundational contribution of these risk genes to ASD.
References:
1. Malwane MI, Nguyen EB, Trejo S Jr, Kim EY, Cucalón-Calderón JR. A Delayed Diagnosis of Autism Spectrum Disorder in the Setting of Complex Attention Deficit Hyperactivity Disorder. Cureus. 2022;14(6):e25825. doi:10.7759/cureus.25825
2. Lai MC, Lombardo MV, Baron-Cohen S. Autism. Lancet. 2014;383(9920):896-910. doi:10.1016/S0140-6736(13)61539-1
3. American Psychiatric Association. DSM-5 Classification. American Psychiatric Association; 2016. https://play.google.com/store/books/details?id=dKTuzgEACAAJ.
4. Loomes R, Hull L, Mandy WPL. What Is the Male-to-Female Ratio in Autism Spectrum Disorder? A Systematic Review and Meta-Analysis. J Am Acad Child Adolesc Psychiatry. 2017;56(6):466-474. doi:10.1016/j.jaac.2017.03.013
5. Thapar A, Rutter M. Genetic Advances in Autism. J Autism Dev Disord. 2021;51(12):4321-4332. doi:10.1007/s10803-020-04685-z
6. Dutheil F, Comptour A, Morlon R, et al. Autism spectrum disorder and air pollution: A systematic review and meta-analysis. Environ Pollut. 2021;278:116856. doi:10.1016/j.envpol.2021.116856
7. Rutter ML. Progress in Understanding Autism: 2007–2010. J Autism Dev Disord. 2011;41(4):395-404. doi:10.1007/s10803-011-1184-2
8. De Rubeis S, He X, Goldberg AP, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515(7526):209-215. doi:10.1038/nature13772
9. Wang N, Lv L, Huang X, et al. Gene editing in monogenic autism spectrum disorder: animal models and gene therapies. Front Mol Neurosci. 2022;15:1043018. doi:10.3389/fnmol.2022.1043018
10. Kizner V, Naujock M, Fischer S, et al. CRISPR/Cas9-mediated Knockout of the Neuropsychiatric Risk Gene KCTD13 Causes Developmental Deficits in Human Cortical Neurons Derived from Induced Pluripotent Stem Cells. Mol Neurobiol. 2020;57(2):616-634. doi:10.1007/s12035-019-01727-1
11. Horie K, Inoue K, Suzuki S, et al. Oxytocin receptor knockout prairie voles generated by CRISPR/Cas9 editing show reduced preference for social novelty and exaggerated repetitive behaviors. Horm Behav. 2019;111:60-69. doi:10.1016/j.yhbeh.2018.10.011
12. Ban Y, Yu T, Wang J, et al. Mutation of the murine Prickle1 (R104Q) causes phenotypes analogous to human symptoms of epilepsy and autism. Exp Neurol. 2022;347:113880. doi:10.1016/j.expneurol.2021.113880
13. Jiang F, Doudna JA. CRISPR-Cas9 Structures and Mechanisms. Annu Rev Biophys. 2017;46:505-529. doi:10.1146/annurev-biophys-062215-010822
14. Pena SA, Iyengar R, Eshraghi RS, et al. Gene therapy for neurological disorders: challenges and recent advancements. J Drug Target. 2020;28(2):111-128. doi:10.1080/1061186X.2019.1630415
15. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816-821. doi:10.1126/science.1225829
16. Xue C, Greene EC. DNA Repair Pathway Choices in CRISPR-Cas9-Mediated Genome Editing. Trends Genet. 2021;37(7):639-656. doi:10.1016/j.tig.2021.02.008
17. Hsieh-Feng V, Yang Y. Efficient expression of multiple guide RNAs for CRISPR/Cas genome editing. aBIOTECH. 2020;1(2):123-134. doi:10.1007/s42994-019-00014-w
Variables
Molar ratio optimization:
Independent variable: molar ratio of backbone plasmids to inserts at 1:50, 1:100, 1:200
Dependent variable: number of clones obtained during bacteria transformation
Controlled variable: reagents, bacteria cultures
Confounding variable: contamination and the quality of reagents
Procedure
Ten different knockout plasmids were constructed in this study. The genes targeted are: EMX2, FGFR1, GLI2, HDAC8, NDPC, PTCH1, RORB, GLI3, POU3F3, SLCLA2. They have been selected due to their potential role in the development of Autism Spectrum Disorder.
Backbone plasmid design and construction
The backbone plasmid used in this project is pAAV-CAG-tdT-2gRNA and is illustrated in figure 1. This plasmid was designed using SnapGene (www.snapgene.com). The backbone plasmid contains a gene encoding the fluorescent protein tdTomato that can be expressed by a CAG promoter and is followed by a poly-A tail, a human U6 promoter for gRNA 1 and a mouse U6 promoter for gRNA 2. The restriction enzyme for gRNA 1 insertion was BsaI and for gRNA 2 insertion was BbsI. There are 2 restriction sites for Bsal with one downstream of the human U6 promoter and one in the ampicillin resistance gene. There are also 2 restriction sites for BbsI downstream of the mouse U6 promoter and one within the gene encoding tdTomato.
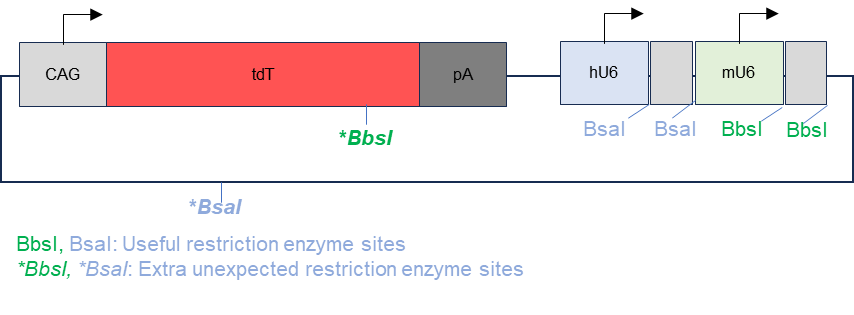
Figure 1. Map of original plasmid with extra restriction enzyme sites. The BsaI and BbsI restriction sites were intended to allow for the insertion of gRNA 1 and gRNA 2. However, the restriction sites were also present on the fluorescent protein tdTomato and the ampicillin resistance gene.
Mutagenesis using PCR was performed on the original plasmid at the BbsI restriction site in tdTomato and at the BsaI restriction site on the ampicillin resistance gene as shown in figure 2 to remove the extra restriction sites. 2 sets of primers, each having a forward and reverse primer, were used to generate a mutation at the restriction site and amplify the corresponding sequences. Gel electrophoresis was conducted afterwards to separate the correct DNA fragments from the original plasmid and the fragments were recovered from the gel through gel recovery using Zymoclean Gel DNA Recovery kit (Zymo ResearchTM, Irvine, California) . The two fragments recovered were combined through homologous recombination. E.coli DH5a was transformed with the final plasmid and the recombinant bacteria went through miniprep using the ZymoPURE Plasmid Miniprep Kit (Zymo ResearchTM, Irvine, California) to extract and purify the backbone plasmids. To validate the final backbone plasmid, restriction digestion with BbsI was used to verify the mutation and the final result was verified through gel electrophoresis.
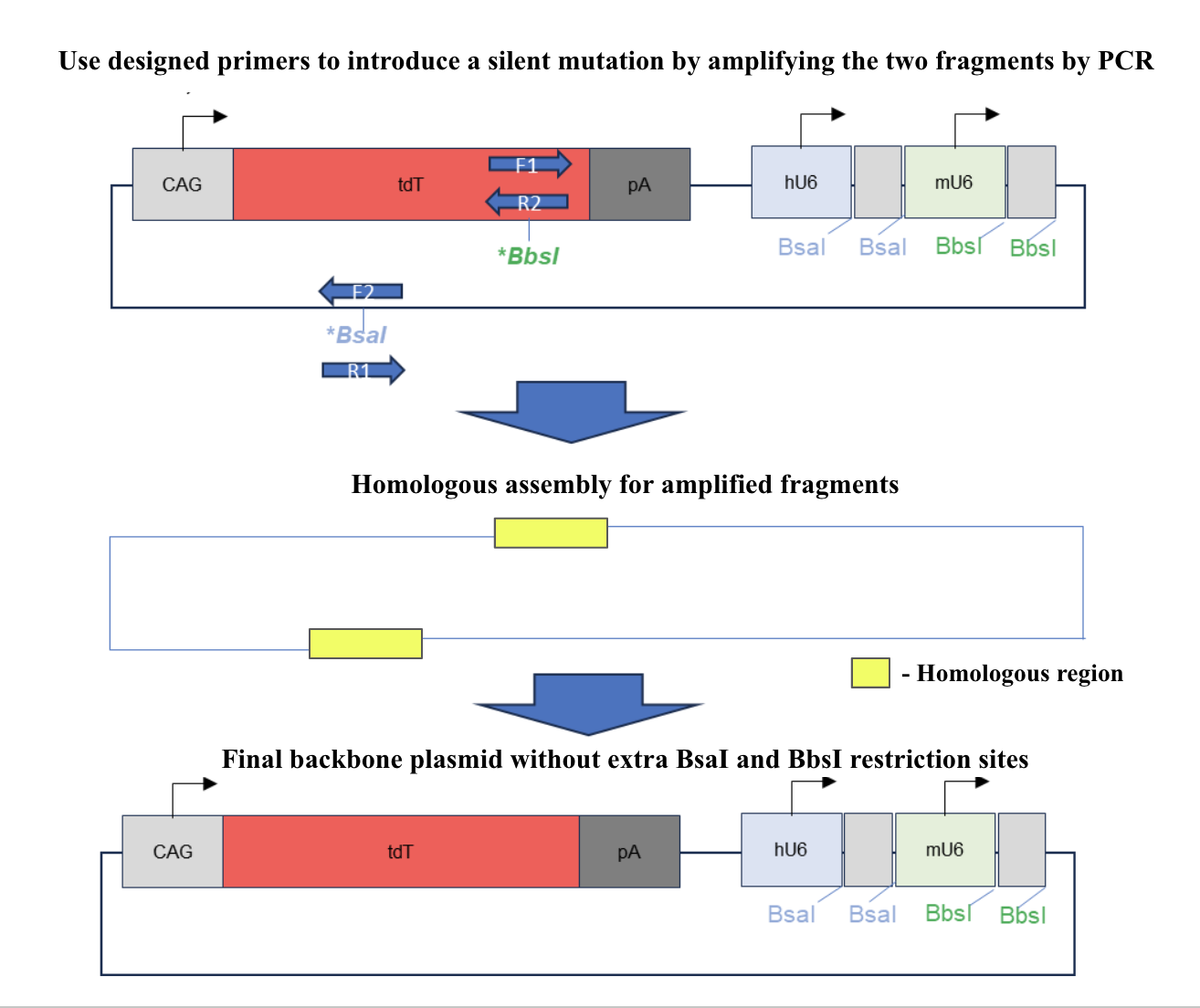
Figure 2. Illustration of mutagenesis for the final backbone plasmid. Forward and reverse primers were used to induce silent mutations and amplify the correct sequence. The amplified, mutated segments were assembled through homologous recombination. The final backbone plasmid has no restriction sites for BsaI and BbsI in places other than the gRNA expression cassettes.
Design of DNA oligos of gRNA sequences
The gRNA primers were designed using CRISPOR.org (http://crispor.org/) and their sequences are shown in Table 1. For each gene, 2 sets of gRNA primers were designed with each set of primers having a forward and reverse primer.
Table 1. Sequences of single-strand DNA oligos used. The sequences of the gRNA oligos shown in the table below were designed using CRISPOR.org (http://crispor.org/).
|
Name |
Sequence |
Name |
Sequence |
|
g1-Emx2_F |
caccgGATGACCCAGATATCGGTAG |
g1-Emx2_R |
aaacCTACCGATACTGGGTCATCcaa |
|
g2-Emx2_F |
ttgtttgCAGGGGCGTCTACTCCAACC |
g2-Emx2_R |
aaacGGTTGGAGTAGACGCCCCTcaa |
|
g1-Fgfrl_F |
cacgTGGAGTTAATACCACCACA |
g1-Fgfrl_R |
aacTGTCGGTGGTATTAACTCCAc |
|
g2-Fgfrl_F |
ttgtttgGCATCGTGGAGAATGAGTAT |
g2-Fgf1_R |
aaacATACTCATTCTCCACGATGCcaa |
|
g1-Gli2_F |
cacgGGGGACCTICTAATGCAGAG |
g1-Gli2_R |
aaaCTCTGCATTAGAAGGTCCCC |
|
g2-Gli2_F |
ttgtttgTACCTGTGTCAGTCCAAGCG |
g2-Gli2_R |
aaaCGCTTGGACTGACACAGGTAcaa |
|
g1-Hdac8_F |
caccgGTTGAGATAGGCATCAGTG |
g1-Hdac8_R |
aacCACTGATGCCTATCTCAACc |
|
g2-Hdac8_F |
ttgtttgCCGTTTGGGGA CCTTCACAA |
g2-Hdac8_R |
aaacTTGTGAAGGTCCCCAAACGGcaa |
|
g1-Ndp_F |
caccgACAGGCTGAAGGACACCAA |
g1-Ndp_R |
aaacTTGGTGTCCTTCAGCACTGTc |
|
g2-Ndp_F |
ttgtttgACTGTACAAATGTAGCTCAA |
g2-Ndp_R |
aacTTGAGCTACATTTGTACAGTcaa |
|
g1-Ptch1_F |
cacgAGCTAATCTCAGACCAACG |
g1-Ptch1_F |
aaaCGTTGGTCTCGAGATTAGCTc |
|
g2-Ptch1_F |
ttgtttgCTGGCAGAGGACTTACGTGG |
g2-Ptch1_R |
aaacCCACGTAAGICCTCTGCCAGcaa |
|
g1-Rorb_F |
cacgGGAGACATGTCAGTACACCA |
g1-Rorb_R |
aacTGGTGTACTGACATTCTCC |
|
g2-Rorb_F |
ttgtttgACAAGTTGGGTACAGATGTG |
g2-Rorb_R |
aaacCACATCTTACCCAACTTGTcaa |
|
g1-GIi3_F |
cacgATACTCGGGCTACTAGATA |
g1-GIi3_R |
aaacTATCTAGTAGCCCGACGTATc |
|
g2-Gli3_F |
ttgttgCCTCGACGTCTAGTGATGAG |
g2-GIi3_R |
aaacCTCATCACTAGACGTCGAGG |
|
g1-Pou3f3_F |
caccgGTCCCCGCGCACTAGCCCG |
g1-Pou3f3_R |
aaacCGGGCTAGTGCGCGGGGACAc |
|
g2-Pou3f3_F |
ttgtttgCAGGGACCCCCCATCACGG |
g2-Pou3f3_R |
aaacCCGTGATGCGGGGGTCCCTGcaa |
|
g1-SIcla2_F |
caccgCATGTTGATAGCCTTCCCGG |
g1-SIcla2_R |
aaacCCGGGAAGGCTATCAACATGa |
|
g2-SIcla2_F |
ttgtttgCCATAGCTCTCGTGCCTAGG |
g2-SIcla2_R |
aaacCCTAGGCACGAGAGGTAcaa |
Molar Ratio of gRNA Insertions
To determine the ideal molar ratio of DNA backbone to gRNA inserts, 1:200, 1:100, 1:50 dilutions of annealed DNA oligos (inserts) were ligated with 50 ng backbone fragments. The optimal molar ratio of insert to backbone DNA fragment was determined to be 1 μL annealed and phosphorylated DNA oligos (1:100) + 50 ng backbone DNA fragment. This ratio was used for the rest of the procedures.
gRNA 1 Insertion and Validation
The backbone plasmids were digested using BsaI and rCutSmart® Buffer (New England BiolabsTM, Ipswich, Massachusetts) at 37°C for 3 hours. To prepare for the insertion of gRNA1, gRNA1 primers were phosphorylated using T4 polynucleotide kinase (FroggaBioTM, Concord, Ontario) and annealed using ramp-down PCR cycling with the following parameters: 37°C for 1 hour, then 95°C for 5 minutes and ramp down to 25°C at 5°C/minute. The inserts were then assembled into the backbone vector using T4 ligase by incubating at 16°C overnight. E. coli DH5a is transformed with the recombinant plasmids and grown on LB agar plates. To validate the insertion of gRNA1, the human U6 forward primer and gRNA 1 reverse primers were used to amplify the inserted gRNA 1 sequence of the clones by PCR. The PCR products were then verified using gel electrophoresis to confirm the correct size of the gRNA1 inserts. The positive plasmids containing the correct size gRNA1 were extracted and purified using ZymoPURE Plasmid Miniprep Kit (Zymo ResearchTM, Irvine, California).
gRNA 2 Insertion and Validation
The recombinant plasmids already containing gRNA1 digested using BbsI and rCutSmart® Buffer (New England BiolabsTM, Ipswich, Massachusetts) at 37°C for 3 hours. The digested plasmids then underwent gel electrophoresis to ensure that the plasmids have gRNA 1 insertion. Plasmids with the correct sequences were recovered from the gel using Zymoclean Gel DNA Recovery kit (Zymo ResearchTM, Irvine, California) . To prepare for the insertion of gRNA2, gRNA 2 primers were phosphorylated using T4 polynucleotide kinase (FroggaBioTM, Concord, Ontario) and annealed using ramp-down PCR cycling method with the following parameters: 37°C for 1 hour, then 95°C for 5 minutes and ramp down to 25°C at 5°C/minute. The inserts were then assembled into the plasmids with gRNA1 insertions using T4 ligase by incubating at 16°C overnight. The plasmids were transformed into E. coli DH5a and grown on LB agar plates. PCR amplification using the human U6 forward primer and gRNA2 reverse primer was performed to validate that the final plasmid contains the insertion of gRNA 1 and 2 as shown in figure 3. The PCR products were then verified using gel electrophoresis.
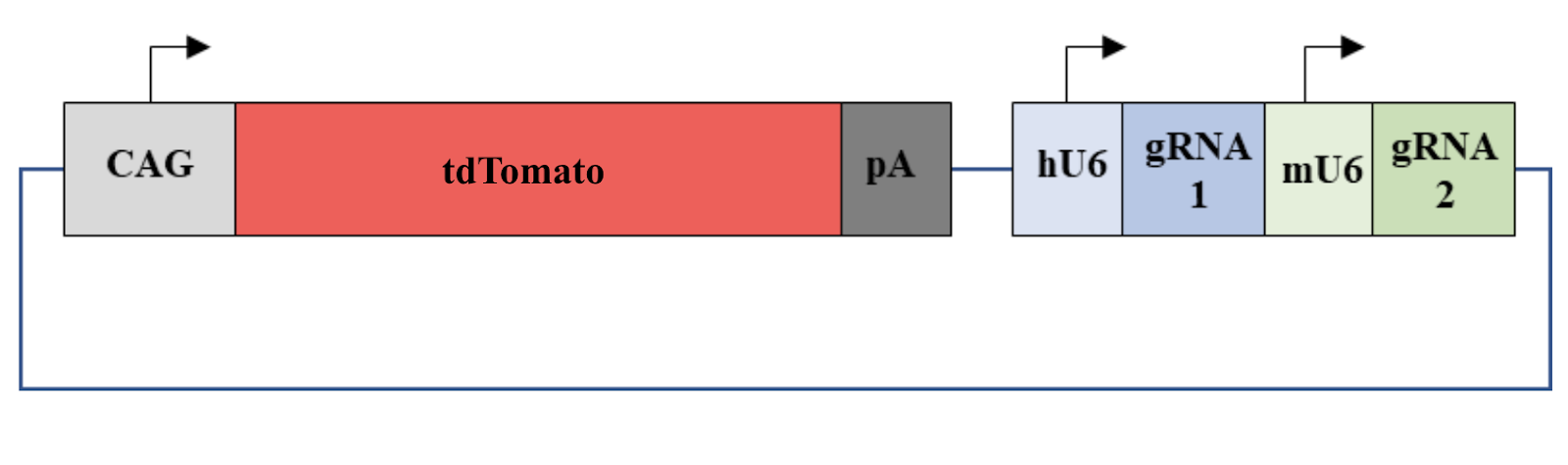
Figure 3. Illustration of the final plasmid. The fluorescent protein tdTomato can be expressed by a CAG promoter. gRNA 1 can be expressed by a human U6 promoter and gRNA 2 can be expressed by a mouse U6 promoter.
Sequencing and Maxiprep
The final plasmids were validated using Sanger sequencing to confirm the correct sequence of their gRNA 1 and gRNA 2 insertions was present in the selected clones. The final plasmids with the correct sequences were extracted from the bacteria clones using ZymoPURE™ II Plasmid Maxiprep Kit (Zymo ResearchTM, Irvine, California).
Observations
1. The effect of different molar ratios of the backbone plasmids to gRNA oligo inserts on the number of clones obtained during bacteria transformation.
|
Molar Ratio |
# of bacteria clones obtained |
|
1:50 |
62 |
|
1:100 |
102 |
|
1:200 |
29 |
2. Gel electrophoresis for the original and backbone plasmids to confirm the elimination of extra restriction sites for the backbone plasmids.

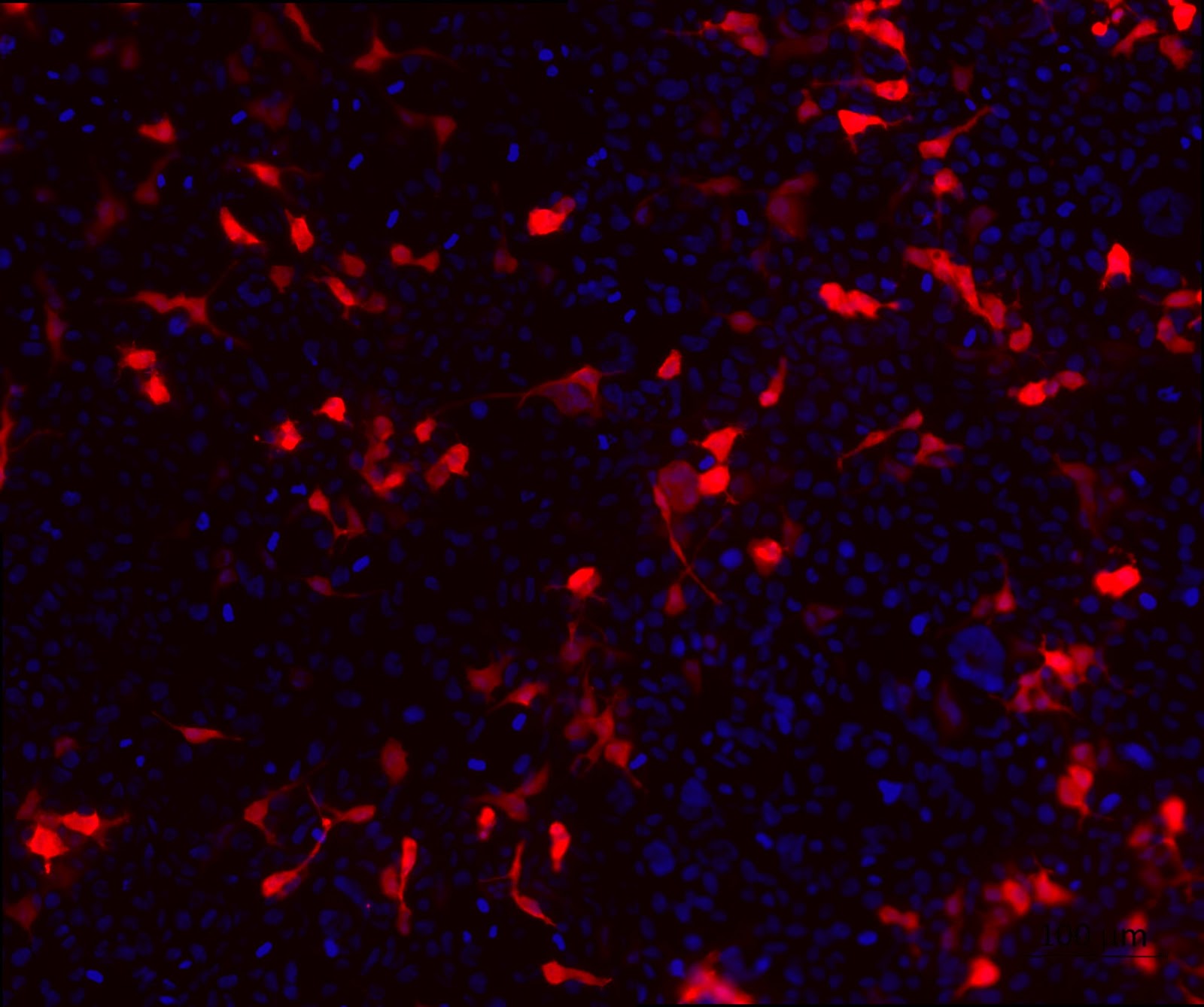
3. HEK cell transfection with modified backbone plasmids to look at the expression of tdTomato.
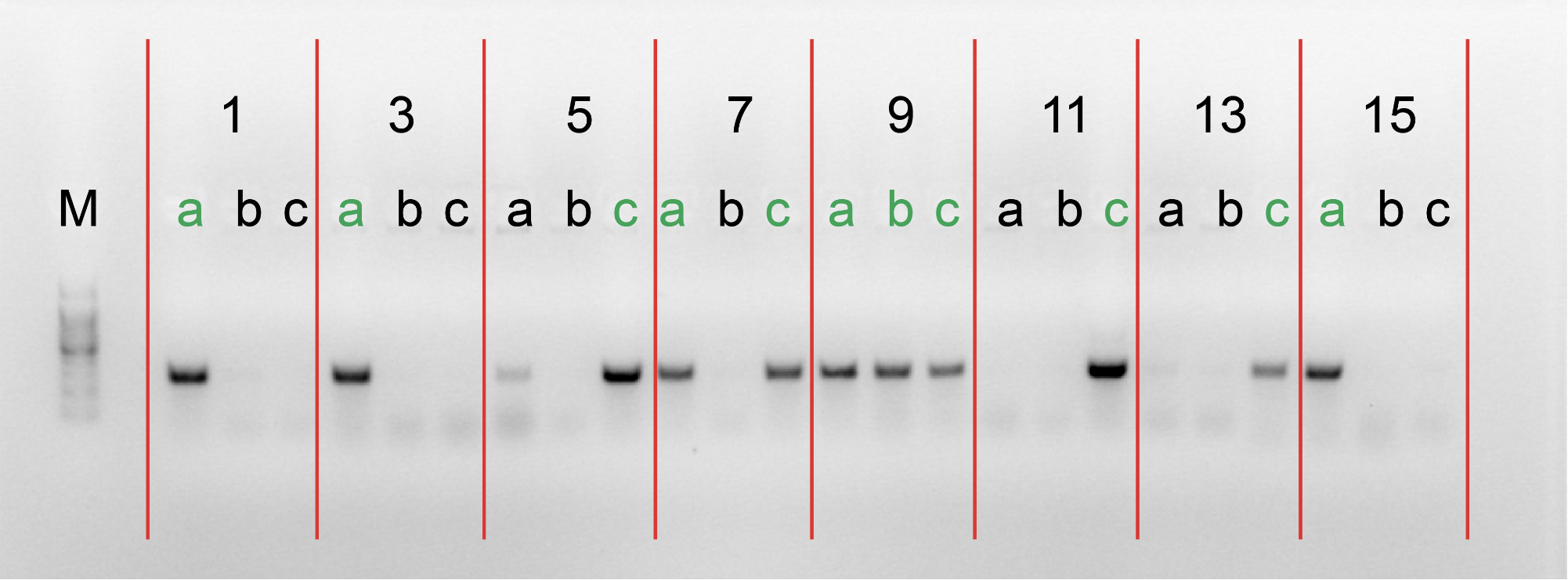
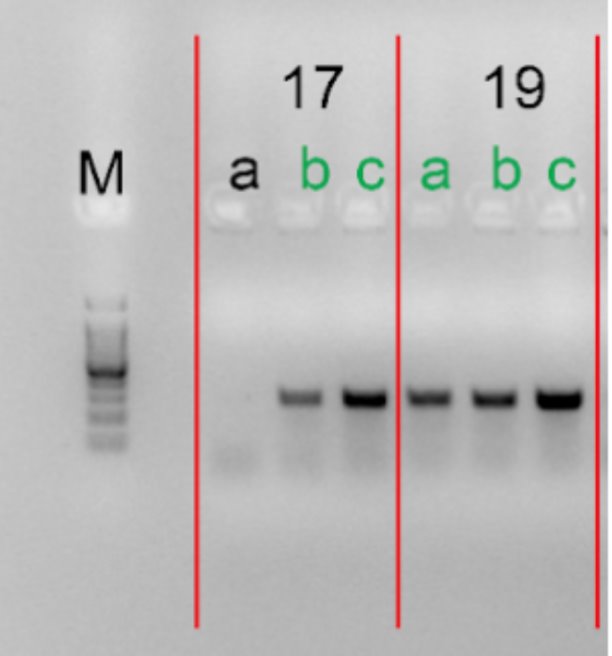
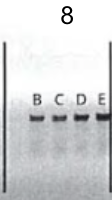
4.Gel electrophoresis after the PCR amplification of gRNA1 expression cassette of the plasmids for the 10 genes
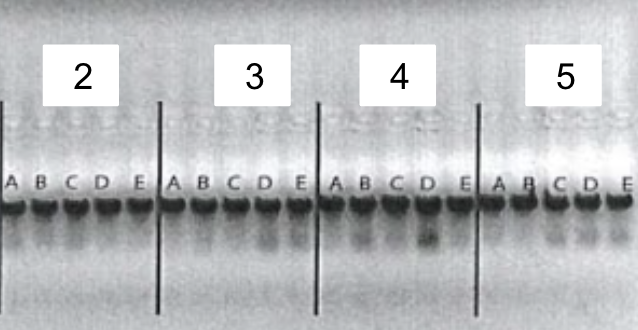
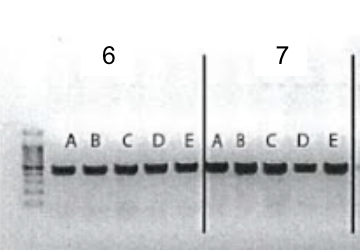
5. Gel electrophoresis after the PCR amplification of gRNA1 and 2 expression cassette of the plasmids for the 10 genes



6. Sanger sequencing results for the plasmid containing the gRNA expression cassettes of EMX2.

Analysis
Backbone Plasmid Optimization
Gel electrophoresis was performed to confirm the sizes of the fragments of the plasmid after restriction enzyme digestion with BbsI and BsaI. From figure 1, which shows the sizes of the different backbone plasmids fragments after digestion with BbsI and BsaI, it can be seen that the mutagenesis was successful since extra restriction sites were eliminated for the modified plasmids, which is indicated by the presence of less bands for the modified plasmids.

Figure 1. Gel electrophoresis for the original and backbone plasmids to confirm the elimination of extra restriction sites for the backbone plasmids. Gel electrophoresis was performed to confirm the number of restriction sites for the modified plasmids. A DNA ladder was used in a lane labeled “M” and plasmids, including original plasmids used as a negative control were digested with BbsI and BsaI.
To confirm the expression of the reporter gene that codes for the fluorescent protein tdTomato as well as the purity of the backbone plasmids, a cell transfection was performed. From figure 2 which shows the expression of tdTomato, it can be seen that the transfection was successful and the quality of the plasmids are fit for mammalian cell transfection.
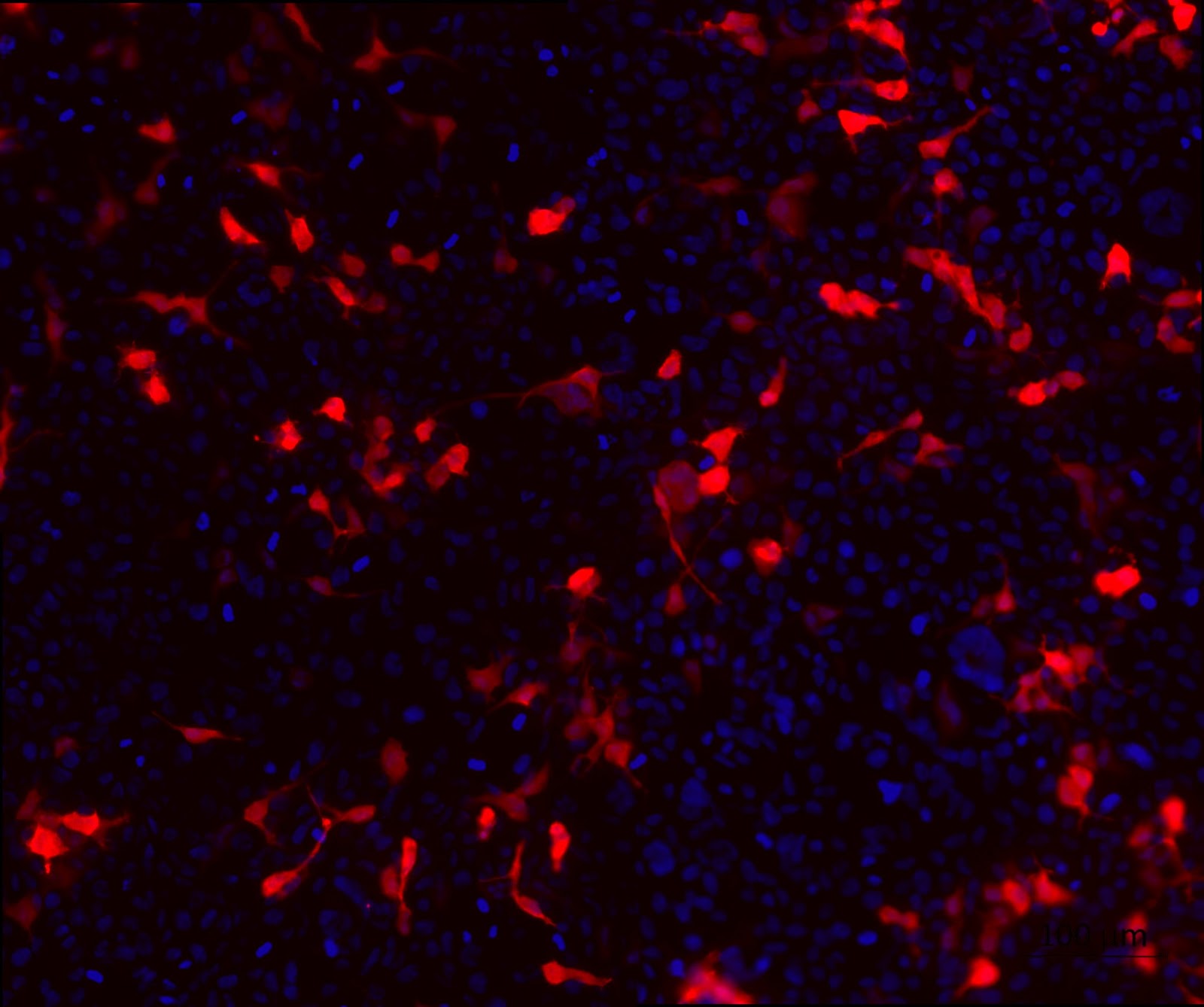
Figure 2. HEK cell transfection with modified backbone plasmids to look at the expression of tdTomato. HEK cells were transfected with HEK 293T cells were transfected with the modified backbone plasmids and fixed using 4% PFA at 24 hours post transfection. tdTomato (red) expression was detected using a fluorescent microscope. Nuclei were stained with DAPI (blue).
After the insertion of gRNA1 oligos, a gel electrophoresis was performed to examine the product of the PCR amplification of the gRNA1 cassettes. Figure 3 shows the result of gel electrophoresis, where the presence of a band of around 260 bp indicates that the amplification was successful.
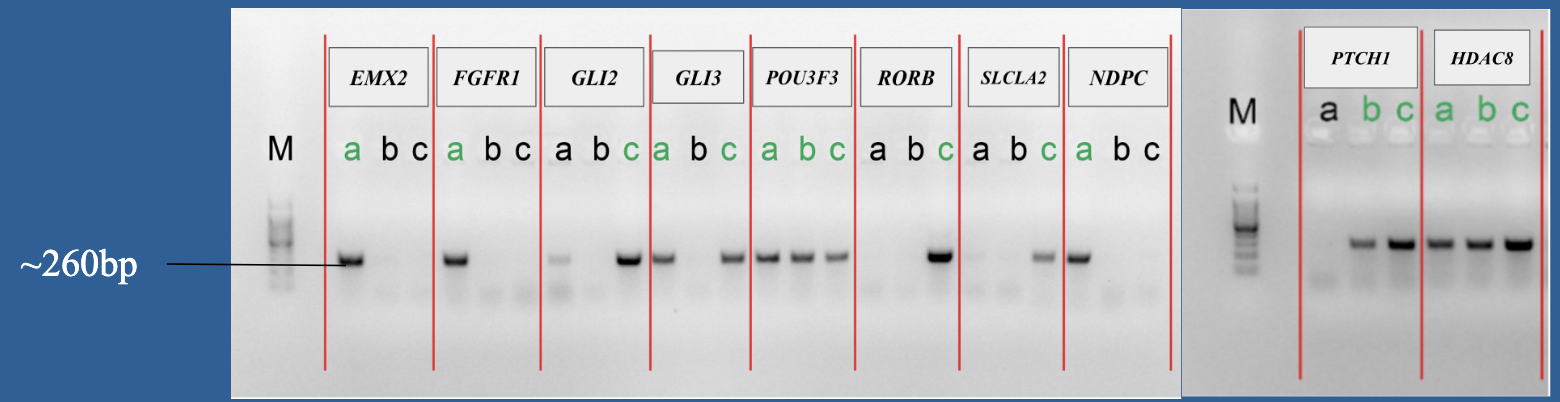
Figure 3. Gel electrophoresis after the PCR amplification of gRNA1 expression cassette of the plasmids for the 10 genes. To examine the result of the PCR amplification, gel electrophoresis was performed to confirm the presence of amplified fragments, which are indicated by bands of around 260 bp. The gene names are included in the boxes above the lanes. Letters (a, b, c) represent clones and positive clones are labeled in green.
After the insertion of gRNA2 oligos, a gel electrophoresis was performed to examine the product of the PCR amplification of the gRNA1 and 2 cassettes. Figure 4 shows the result of gel electrophoresis, where the presence of a band of around 450 bp indicates that the amplification was successful.
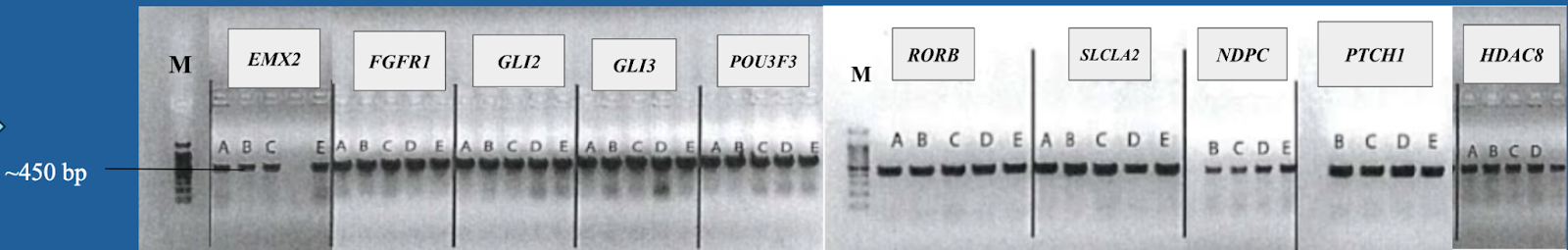
Figure 4. Gel electrophoresis after the PCR amplification of gRNA1 and 2 expression cassette of the plasmids for the 10 genes. To examine the result of the PCR amplification, gel electrophoresis was performed to confirm the presence of amplified fragments, which are indicated by bands of around 450 bp. The gene names are included in the boxes above the lanes. Positive clones are labeled by letters (A, B, C, D, E).
Sanger sequencing was performed to validate the exact sequence of the final plasmids. Figure 5 shows the Sanger sequencing result for the plasmid of EMX2. From the results, it can be seen that gRNA1 and 2 cassettes were successfully inserted into the backbone plasmids.

Figure 5. Color-coded result of Sanger sequencing for the plasmid containing gRNAs of EMX2. Sanger sequencing was performed to confirm the sequence of the final plasmids. The hU6 promoter is followed by gRNA1 and the mU6 promoter is followed by gRNA2.
Conclusion
-
Knockout plasmids for 10 ASD-risk genes are successfully constructed using restriction enzyme and T4 DNA ligase based cloning method as well as homologous-recombination based assembly
-
Mutagenesis on the backbone plasmid was successful
-
The ideal molar ratio of backbone plasmid to annealed inserts has been determined to be 1:100
-
Bacteria clones are ready for Maxiprep to be used in in vivo study
Application
-
Make the current KO plasmids more effective: utilizes 2 gRNA instead of 1 to double the success rate of gene knockout
-
2 gRNA system can revolutionize KO plasmid system for research in other fields, e.g., cancer research, polygenic disorders, and genetic metabolic disorders
-
Knockout plasmids for 10 not well-studied ASD-risk genes are established; allows for the establishment of mouse models
-
Further advance the current understanding of the roles of the following ASD-risk genes: EMX2 (Empty Spiracles Homebox 2), FGFR1 (Fibroblast Growth Factor Receptor 1), GLI2 (GLI Family Zinc Finger 2), GLI3 (GLI Family Zinc Finger 3), POU3F3 (POU Class 3 Homebox 3), RORB (RAR-Related Orphan Receptor B), SLCLA2 (Solute Carrier Family 1 Member 2), NDPC (Neurogenic differentiation I), PTCHI (Patched I), HDAC8 (Histone Deacetylase 8).
In a future project:
-
The knockout plasmids will be packaged into Adeno-associated virus (AAV)
-
Mice will be infected with the virus containing ASD knockout plasmid
-
The Cas9 expression in these mice will be under the control of a Cre-Lox system
-
The role of the ASD risk genes will be evaluated in astrocytes by perturbation of the genes
-
The mice will be used for single cell RNA-seq and behavioral study of ASD symptoms
Sources Of Error
-The experiment to determine the ideal molar ratio could have been conducted for more trials
-Potential contaminations in the reagents
Citations
References:
1. American Psychiatric Association. DSM-5 Classification. American Psychiatric Association; 2016. https://play.google.com/store/books/details?id=dKTuzgEACAAJ.
2. Ban Y, Yu T, Wang J, et al. Mutation of the murine Prickle1 (R104Q) causes phenotypes analogous to human symptoms of epilepsy and autism. Exp Neurol. 2022;347:113880. doi:10.1016/j.expneurol.2021.113880
3. Concordet JP, Haeussler M. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018;46(W1):W242-W245. doi:10.1093/nar/gky354
4. De Rubeis S, He X, Goldberg AP, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515(7526):209-215. doi:10.1038/nature13772
5. Dutheil F, Comptour A, Morlon R, et al. Autism spectrum disorder and air pollution: A systematic review and meta-analysis. Environ Pollut. 2021;278:116856. doi:10.1016/j.envpol.2021.116856
7. Horie K, Inoue K, Suzuki S, et al. Oxytocin receptor knockout prairie voles generated by CRISPR/Cas9 editing show reduced preference for social novelty and exaggerated repetitive behaviors. Horm Behav. 2019;111:60-69. doi:10.1016/j.yhbeh.2018.10.011
8. Jiang F, Doudna JA. CRISPR-Cas9 Structures and Mechanisms. Annu Rev Biophys. 2017;46:505-529. doi:10.1146/annurev-biophys-062215-010822
9. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816-821. doi:10.1126/science.1225829
10. Kizner V, Naujock M, Fischer S, et al. CRISPR/Cas9-mediated Knockout of the Neuropsychiatric Risk Gene KCTD13 Causes Developmental Deficits in Human Cortical Neurons Derived from Induced Pluripotent Stem Cells. Mol Neurobiol. 2020;57(2):616-634. doi:10.1007/s12035-019-01727-1
11. Lai MC, Lombardo MV, Baron-Cohen S. Autism. Lancet. 2014;383(9920):896-910. doi:10.1016/S0140-6736(13)61539-1
12. Loomes R, Hull L, Mandy WPL. What Is the Male-to-Female Ratio in Autism Spectrum Disorder? A Systematic Review and Meta-Analysis. J Am Acad Child Adolesc Psychiatry. 2017;56(6):466-474. doi:10.1016/j.jaac.2017.03.013
13. Malwane MI, Nguyen EB, Trejo S Jr, Kim EY, Cucalón-Calderón JR. A Delayed Diagnosis of Autism Spectrum Disorder in the Setting of Complex Attention Deficit Hyperactivity Disorder. Cureus. 2022;14(6):e25825. doi:10.7759/cureus.25825
14. Pena SA, Iyengar R, Eshraghi RS, et al. Gene therapy for neurological disorders: challenges and recent advancements. J Drug Target. 2020;28(2):111-128. doi:10.1080/1061186X.2019.1630415
15. Rutter ML. Progress in Understanding Autism: 2007–2010. J Autism Dev Disord. 2011;41(4):395-404. doi:10.1007/s10803-011-1184-2
16. Thapar A, Rutter M. Genetic Advances in Autism. J Autism Dev Disord. 2021;51(12):4321-4332. doi:10.1007/s10803-020-04685-z
17. Wang N, Lv L, Huang X, et al. Gene editing in monogenic autism spectrum disorder: animal models and gene therapies. Front Mol Neurosci. 2022;15:1043018. doi:10.3389/fnmol.2022.1043018
18. Xue C, Greene EC. DNA Repair Pathway Choices in CRISPR-Cas9-Mediated Genome Editing. Trends Genet. 2021;37(7):639-656. doi:10.1016/j.tig.2021.02.008
Acknowledgement
I would like to thank Dr. Garcia-Diaz for her guidance and Dr. Lizheng Wang and Dr. Jiami Guo for their supervision in project design and experiments.